

Ijraset Journal For Research in Applied Science and Engineering Technology
Drug Target Interaction [DTI] and Prediction using Machine Learning
Authors: Tejas Kumar M, Rakesh M D
DOI Link: https://doi.org/10.22214/ijraset.2022.46901
Certificate: View Certificate
Abstract
The need to find new antibiotics is expanding as a result of the quick rise in bacteria that are resistant to medicines. Discovering drug-protein interactions could be an essential first step in the process of developing drugs since it will substantially reduce the scope of the look for possible solutions. Since in vitro assays are extremely time-consuming and pricey. We developed a machine learning method that can predict medications for the target in order to overcome this difficulty. We used the Padel script to do predictions on several chemical libraries, acquire drug physical and chemical properties, and obtain features extracted. establishing which model is best for predicting drug-target interactions is performed by analyzing the Random Forest technique with the Naive Bayes method, K-Nearest Neighbor, and other choices. This study reduces the failure rates and costs incurred when creating new pharmaceuticals while demonstrating the value of adopting machine learning approaches in drug discovery.
Introduction
I. INTRODUCTION
As a greater number of drugs become ineffective against the bacteria, the prevalence of resistant bacteria is becoming a growing concern for both the general public and the pharmaceutical business. Despite the fact that antibiotic therapy is in line with modern medicine, a decline in funding makes it difficult for investigators to stay informed of the actual population's healthcare needs Aslam et al. (2018) [1]. Traditional drug discovery takes a long and is exorbitant; for example, in 2006, the Food and Drug Administration (FDA) only approved 22 potential biological entities in spite of enormous research and development costs of up to $93 billion USD Yu et al. (2012) [2]. One of the key aspects of drug identification is the determination of interactions between compounds and proteins. Therefore, there is a tremendous motivation to create novel techniques that can quickly identify these possible drug-protein interactions Yamanishi et al. (2009) [3].
Maximum techniques were developed to evaluate and estimate molecule-protein interactions. Approaches based on chemicals and docking are two of the most common. The underpinning of ligand-based strategies is the theory that substances with similar abilities ought to be bound to the identical category of molecule Keiser et al. (2008) [4] invented the Ensemble Approach a mechanism of quantitatively related receptors (proteins) based on the protein similarity with their ligands.. However, when there are enough known major ingredients for a target of interest, the performance of the ligand-based strategy is usually substandard. Another widely used approach is the Docking Simulation approach which help for structure-based drug design Tian et al. (2016) [6], Utilizing three-dimensional objects and molecular docking, Li et al. (2006) [7] developed a useful tool for target identification, TarFisDock, When a minor material's potential protein targets are determined utilizing reverse ligand-protein docking Yang et al. (2011) [8] established the Chemical-Protein Interactome docking technology in order for replicating diversity in connections between drugs and a variety of human proteins, Unfortunately, it requires more time to finish trial simulation trials since many proteins lack three-dimensional structures. Chemogenomic methods were used increasingly commonly than the classic demand for product methods as a result of the increase in biological and chemical data available for prediction. Yamanishi et al. (2009) [3] an uniform space known as the pharmacological space it incorporates the chemical form and the genomic form to infer DTIs, In this proposed method, Chemical space refers to the variety of specific chemical compounds' chemical structures that are similar, genomic space relates to the spectrum of possible proteins' amino acid sequences that become similar, and pharmacological space refers to the range of interactions that reflect the network of interactions between drugs and their goals. Recent advancements in machine learning enhance their capacity to identify connections and patterns among the information connected to drugs and targets. Cao et al. (2014) [10] combined chemical data, Molecular Access System (MACCS) fingerprints and/or biological information, protein descriptors, network characteristics, and substructure fingerprints are combined to create feature vectors that can be employed in a predictive random forest (RF) model, to identify new DTIs. Nagamine et al.
(2007) [11] used a support vector machine as the drug-protein model to infer new interactions. yamanishi et al. (2009) [3] devised a method for supervised prediction utilizing bipartite local models, one based on protein resemblance and the other on elemental composition similarity.
In this work, we propose a machine learning method for the prediction of Drug Target Interaction using SMILE strings which represent the chemical formula of Drugs and Targets which is taken from the Chembl database. We Investigated four supervised machine learning models: k-nearest neighbors (KNN), Random Forest (RF), and Naïve Bayes, and also, compared the result of three algorithms in terms of Accuracy. we successfully identify that Random Forest provides the best accuracy prediction among all three methods.
II. RELATED WORK
Ruolan Chen et.al, focused on machine learning approaches by summarizing a detailed list of data sets frequently used in drug discovery processes and by applying a classification scheme that is hierarchical and many ideal methods of each and every category are introduced. They have also identified the advantages and disadvantages of approaches in each and every category. Zaynab Mousavian et.al, have provided a useful idea that has emerged in this paper. In post-genomic drug discovery, the extensive combining genomic, proteomic, and signaling data, and metabolomic data may make it possible to build intricate cellular networks. Maryam Bagherian et.al, have explained the data needed for DTIs to foresee are followed by a broad list that includes machine learning approaches and databases, that have been proposed and utilized to foresee DTIs. The main useful features of each set of approaches are also discussed in detail. Heba El-Behery et.al, have proposed the DTIs expected model in this research, which makes use of the special qualities of pharmaceuticals and proteins with a structure. The model is built on the cooperation of learning algorithms to predict DTI and gives better accuracy in results from the data consisting of both structures and its features, as shown by the results of comparing it with various methods that are already in use under K-fold cross-validation.
III. METHODOLOGY
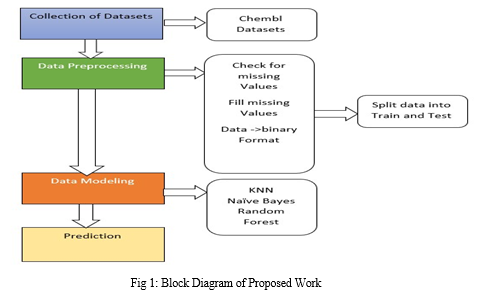
Figure 1 depicts the Block diagram of the proposed system. The Kaggle is the repository which the data sets are collected from. Then preprocessing of data is done with the help of the Padel script. The Pre-processed data is then divided into Train and Test data sets and given to the model. The data obtained is analyzed and predicted. The three algorithms are used for prediction namely Random Forest (RF), K-nearest neighbor(KNN), and Naïve Bayes. The most accurate algorithm can be found for Drug Target Interaction Prediction.
A. Collection of Datasets
Datasets of Chembl Beta-Lactamase are collected and used for further process, the datasets are converted into binary formats with the help of padel script.
B. Data Processing
We will further categorize the datasets into missing and non-missing values based on the datasets, by taking into account the existence or lack of functional values in the database of molecules.
Additionally, non-missing data is separated into active and inactive data.
The molecular value of the chembl datasets shows that the specific value indicates the drug's ability to inhibit the target. Chemical groups whose inhibition values are greater than 5 are listed in the Active group.
The molecular value of the chembl datasets shows that the specific value indicates the drug's ability to inhibit the target. Chemical groups whose inhibition values are less than or equal to 5 are listed in the In-Active group
Furthermore, data is classified into the following steps:
- Split the data into train and test data, to train the mode.
- Y data contains the values based on the p-chembl values (i.e., 0 & 1).
- X data contains the values of molecular structure in the form of binary.
C. Data Modelling
Splited data is applied to the machine learning algorithm.
- Random Forest.
- Naive Bayes.
- K-nearest Neighbor
Algorithms used for the Prediction of Drug Target Interaction.
The algorithm for machine learning is an approach by which the system of AI capabilities performs the processes, normally by foreseeing the values as output from already provided data as input.
D. Random Forest
It is supervised learning that integrates predictions from two or more models and is based on the idea of ensemble learning. It is characterized as a classifier because it averages numerous decision trees on various subsets of the provided data to increase the anticipated accuracy the information set. This combines the results of multiple decision trees to provide a response that reflects the average of all of them. Despite having identical nodes, each of these decision trees uses different data to produce a variety of leaves.[16]
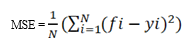
Mean square error (MSE) is used to solve the Random Forest problem, where N denotes the number of data points, fi denotes the output value of the model, and Yi denotes the actual value of the data point [16].
This formula calculates the distance between each node and the expected real value in order to determine which branch is the best option for your forest. In this case, fi is the value the decision tree returned, and Yi is the value of the data point you are testing at a particular node. Random Forest’s key benefits include being used for regression and classification problems to create a diversified model, preventing data overfitting, and being quick to train with test data [16].
E. K-Nearest Neighbor
It ranks among the most fundamental machine learning algorithms that is based on supervised learning. It compiles all of the information available and groups new information according to commonalities. This means that the KNN approach can be used to swiftly and accurately categorize newly generated data. It is mostly used to classify data depending on how its neighbors are classified. The parameter K in KNN denotes the number of closest neighbors to be taken into account for determining the winner by majority vote. The Sqrt(n), where n is the total number of data points, must then be obtained before selecting K. The main advantages of KNN are that they are simple to construct, robust against noisy training data, and can perform better when the training data is vast. [16].
F. Naive Bayes
It ranks among the simplest and most effective classification techniques, facilitating the creation of quick machine learning models that could produce trustworthy predictions. The Bayes' theorem, often known as Bayes' law, is employed to assess the Probability of a given hypothesis with some prior knowledge. Determined by the conditional probability this [17]. The recipe for Bayes’ theorem is given as:
were,
- P(A—B): It’s the probability of hypothesis A on the noticed B event.
- P(B—A): It’s the given data probability that the hypothesis probability is true.
- P(A): It’s the hypothesis probability before noticing the data.
- P(B): proof of the data probability. Naive Bayes’ true advantage is that it is a quick and simple technique to forecast a class of datasets. Both binary and multiclass classifications can be done with it.
IV. IMPLEMENTATION AND RESULTS
A. Plan of Execution
- Using the Kaggle Machine Learning repository which comprises a data set containing Drug data.
- The collected datasets are pre-processed and analyzed using a machine learning library.
- The pre-processed datasets are spitted into training and testing and passed to the machine learning algorithm.
- The trained datasets are compared with test results with help of an algorithm and results are shown in Percentage with a bar graph.
- The results are compared with the applied algorithms and the algorithm showing the best results is considered.
As per the above plan if execution the data sets are taken from the Kaggle repository, then based on the molecular value from the database the drug data are considered and these data are pre-processed. The pre-processed data is divided into Train and Test data sets. In our work, we have considered two combinations one is 70/30 and the other is 80/20 as Train and Test data sets. After applying these two combinations of data sets into the algorithms the one that shows accurate results is considered the best model for the prediction of Drug protein Interaction.
Total number of missing and non-missing values from the database is shown in Figure 3.
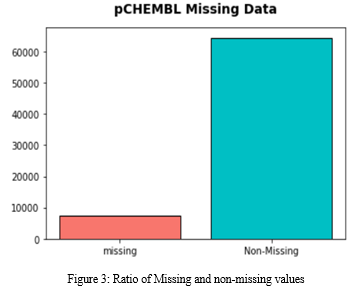
Padel script helps in the conversion of SMILES (chemical representation of drug) into binary formats with the help of fingerprints which acts as a library. after classifying the data into missing and non-missing values the non-missing values are further processed in order to obtain the binary formats of chemical notations which further helps in train and test the machine learning model.

As shown in Figure 9, among the three algorithms Random Forest algorithm gives a better prediction accuracy of 85%, followed by KNN at 76 % and Naïve Bayes at 56%.
B. Comparative study of Applied Algorithms

Table 1, shows the comparison of all three algorithms for two different combinations of data sets percentages such as 70 30 and 80 20 as Train and Test data. By this, we can understand that 80 20 combinations of Train and Test data sets are showing the best results for the Random Forest model with 85.16 % accuracy
V. FUTURE SCOPE
- The Prediction may be expanded further to allow the researcher to see how each interaction turns out.
- By giving researchers the choice of the datasets to be used, we can further enhance performance.
- It is possible to compare a few more algorithms.
Conclusion
In this paper, we investigated a classification system using a new chembl database and extraction of features with the help of padel script. We tested three supervised machine learning models: k-nearest neighbors (KNN), Random Forest (RF), and Naïve Bayes. We tested the performance of these techniques in classifying: test data and train data into 70/30 ratio and 80/20 ratio after pre-processing and extraction of the data and measuring the accuracy. The plot shows that the Random Forest had the best performance in comparison with the other methods by considering the 80/20 ratio.
References
[1] M. Nirmala Devi, S. Mahima, R. Ramupriya, Sumaya Abdul Sathar, \"Improvised CNN model to Predict SARS by Detecting the Localisation of Proteins\", 2021 5th International Conference on Electronics, Communication and Aerospace Technology (ICECA), pp.822-828, 2021. [2] Cao DS, Zhang LX, Tan GS, Xiang Z, Zeng WB, Xu QS, Chen AF. Computational Prediction of Drug-Target Interactions Using Chemical, Biological, and Network Features. Mol Inform. 2014 Oct;33(10):669-81. doi: 10.1002/minf.201400009. Epub 2014 Sep 26. PMID: 27485302. [3] Yu H, Chen J, Xu X, Li Y, Zhao H, Fang Y, Li X, Zhou W, Wang W, Wang Y. A systematic prediction of multiple drug-target interactions from chemical, genomic, and pharmacological data. PLoS One. 2012;7(5):e37608. doi: 10.1371/journal.pone.0037608. Epub 2012 May 30. PMID: 22666371; PMCID: PMC3364341. [4] Nobuyoshi Nagamine, Yasubumi Sakakibara, Statistical prediction of protein–chemical interactions based on chemical structure and mass spectrometry data, Bioinformatics, Volume 23, Issue 15, August 2007, Pages 2004–2012, https://doi.org/10.1093/bioinformatics/btm266 [5] Çobano?lu, Murat & Liu, Chang & Hu, Feizhuo & Oltvai, Zoltan & Bahar, Ivet. (2013). Predicting Drug-Target Interactions Using Probabilistic Matrix Factorization. Journal of chemical information and modeling. 53. 10.1021/ci400219z. [6] Yamanishi Y, Araki M, Gutteridge A, Honda W, Kanehisa M. Prediction of drug-target interaction networks from the integration of chemical and genomic spaces. Bioinformatics. 2008 Jul 1;24(13):i232-40. doi: 10.1093/bioinformatics/btn162. PMID: 18586719; PMCID: PMC2718640. [7] Stokes JM, Yang K, Swanson K, Jin W, Cubillos-Ruiz A, Donghia NM, MacNair CR, French S, Carfrae LA, Bloom-Ackermann Z, Tran VM, Chiappino-Pepe A, Badran AH, Andrews IW, Chory EJ, Church GM, Brown ED, Jaakkola TS, Barzilay R, Collins JJ. A Deep Learning Approach to Antibiotic Discovery. Cell. 2020 Feb 20;180(4):688-702.e13. doi: 10.1016/j.cell.2020.01.021. Erratum in: Cell. 2020 Apr 16;181(2):475-483. PMID: 32084340; PMCID: PMC8349178. [8] Ruolan Chen, Xiangrong Liu, Shuting Jin ,Jiawei Lin,Juan Liu ”Machine Learning for Drug-Target Interaction Prediction ” ,2018 . [9] J., S. K., & S., G. (2019). Prediction of heart disease using machine learning algorithms. 2019 1st International Conference on Innovations in Information and Communication Technology (ICIICT), 1–5. https : / / doi . org / 10 . 1109 / ICIICT1 . 20198741465. [9] Ali Masoudi-Nejad ,Zaynab Mousavian and Joseph H Bozorgmehr ”Drug-target and disease networks: polypharmacology in the post-genomic era ” ,2013 . [10] Maryam Bagherian ,Elyas Sabeti ,Kai Wang, Maureen A. Sartor,Zaneta Nikolovska-Coleska and Kayvan Najarian ”Machine learning approaches and databases for prediction of drug–target interaction ” ,2021 [11] Heba El-Behery a, Abdel-Fattah Attia Nawal El-Fishawy ,Hanaa Torkey ”Efficient machine learning model for predicting drug-target interaction with case study for Covid-19”,2021 . [12] George Adam , Ladislav Rampa´sek, Zhaleh Safikhani, Petr Smirnov , ? Benjamin Haile-Kains1and , Anna Goldenberg ” Machine learning approaches [13] Li, Y., Huang, Y.-A., You, Z.-H., Li, L.-P. & Wang, Z. (2019). Drug-target interaction prediction is based on drug fingerprint information and protein sequence. Molecules, 24(16), 2999. [14] Marcos-Garc?a, J.-A., Mart?nez-Mon ?es, A. & Dimitriadis, Y. (2015). Despro: A method based on roles to provide Collaboration analysis support adapted to the participants in cscl situations. Computers & Education,82, 335–353. [15] Adam, G., Rampášek, L., Safikhani, Z., Smirnov, P., Haibe-Kains, B., & Goldenberg,A. (2020). Machine learning approaches (to drug response prediction: Challenges andrecent progress. NPJ precision oncology, 4(1), 1–10 [16] Patel L, Shukla T, Huang X, Ussery DW, Wang S. Machine Learning Methods in Drug Discovery. Molecules. 2020 Nov 12;25(22):5277. doi: 10.3390/molecules25225277. PMID: 33198233; PMCID: PMC7696134. [17] Kowalewski, J., & Ray, A. (2020). Predicting novel drugs for sars-cov-2 using machine learning from a> 10 million chemical space. Heliyon, 6(8), e04639 [18] Maha A. Thafar, Rawan S. Olaya, Somayah Albaradei ”DTi2Vec: Drug–target interaction prediction using network embedding and ensemble learning ” ,2021 . [19] Iqbal Osisanwo F.Y., Akinsola J.E.T., Awodele O., Hinmikaiye J. , Olakanmi, Akinjobi J. ”Supervised Machine Learning Algorithms: Classification and Comparison ”,2017.
Copyright
Copyright © 2022 Tejas Kumar M, Rakesh M D. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.

Download Paper
Paper Id : IJRASET46901
Publish Date : 2022-09-27
ISSN : 2321-9653
Publisher Name : IJRASET
DOI Link : Click Here
 Submit Paper Online
Submit Paper Online