

Ijraset Journal For Research in Applied Science and Engineering Technology
Formulation and Evaluation of Analytical Methods for Determination of Indacaterol
Authors: Sachin Laxman Ingale , Dr. Hingane L. D., Prof. Nakhate S. T.
DOI Link: https://doi.org/10.22214/ijraset.2022.44995
Certificate: View Certificate
Abstract
Indacaterol is a novel class of ?2 adrenoreceptor -agonist used to treat chronic obstructive pulmonary disease. Various analytical methods used for the estimation of Indacaterol have been reviewed in this project. These methods include Ultraviolet Spectrometric method, High Performance Liquid Chromatography method, Capillary electrophoretic method, TLC densitometric method for qualitative and quantitative estimation of Indacaterol in pharmaceutical formulation
Introduction
I. OBJECTIVE
The objective of the review study is qualitative and quantitative estimation of Indacaterol in bulk drug and pharmaceutical formulation by different anaytical methods such as UV Spectroscopic method, High Performance Liquid Chromatography method, Capillary electrophoretic method, TLC densitometric method.
II. INTRODUCTION
Indacaterol is in a class of medications called long-acting beta agonists (LABAs). It works by relaxing and opening air passages in the lungs, making it easier to breathe.
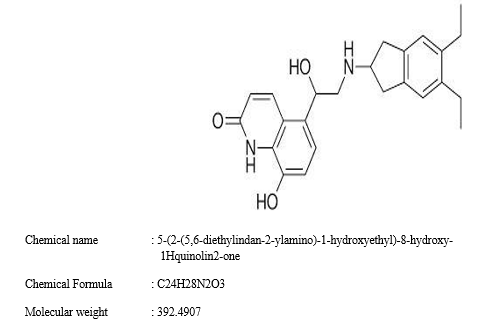
Structure: Indacaterol
Mechanism of Action:
Indacaterol works by stimulating adrenergic beta-2 receptors in the smooth muscle of the airways. This causes relaxation of the muscle, thereby increasing the diameter of the airways, which become constricted in asthma and COPD. It is also long acting due to its high affinity to the lipid raft domains in the airway membrane so it slowly dissociates from the receptors. Indacaterol also has a high intrinsic efficacy so it is also very rapid acting - onset of action occurs within 5 minutes.
III. ANALYTICAL TECHNIQUES FOR ESTIMATION OF INDACATEROL:
- UV Spectroscopic method
- IR Spectroscopy method
- TLC Densitometric method
- High Performance Liquid Chromatography method
- Capillary electrophoretic method
A. UV Spectroscopic Method
Y.A. Salem et al.3 developed Spectroscopic method for estimation of Indacaterol in capsules. Three simple and sensitive spectrophotometric methods (A, B and C) have been developed for the quantitative estimation of Indacaterol (IND) in bulk drug and capsules.
1) Principle
- Basically, spectroscopy is related to the interaction of light with matter.
- As light is absorbed by matter, the result is an increase in the energy content of the atoms or molecules.
- When ultraviolet radiations are absorbed, this results in the excitation of the electrons from the ground state towards a higher energy state.
- Molecules containing π-electrons or non-bonding electrons (n-electrons) can absorb energy in the form of ultraviolet light to excite these electrons to higher anti-bonding molecular orbitals.

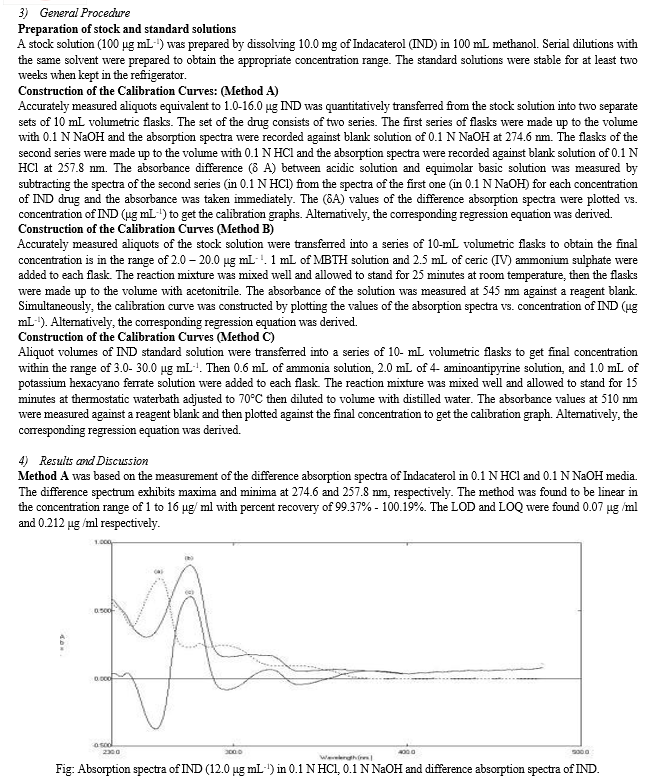
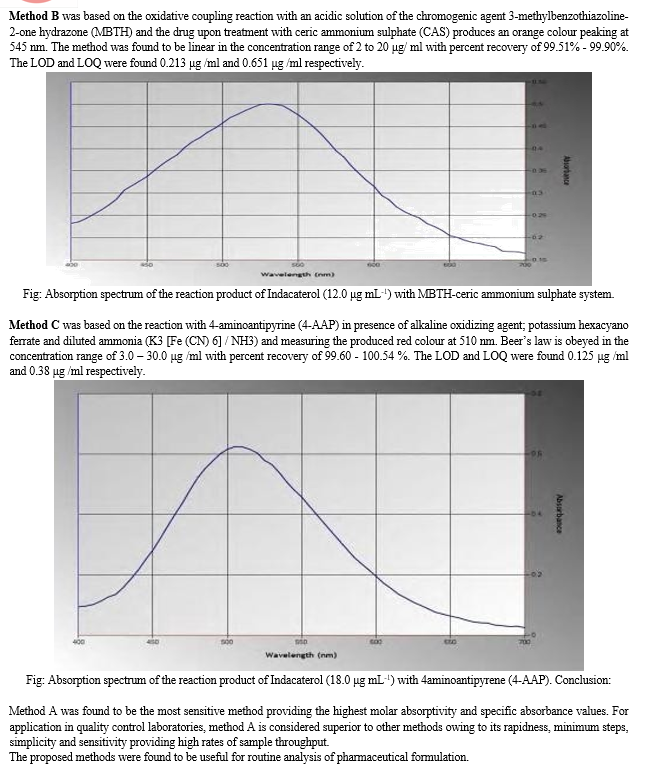
B. Infrared Spectroscopy
Infrared (IR) spectroscopy is one of the most common and widely used spectroscopic techniques employed mainly by inorganic and organic chemists due to its usefulness in determining structures of compounds and identifying them. Chemical properties due to the presence of different functional groups.
IR spectroscopy (which is short for infrared spectroscopy) deals with the infrared region of the electromagnetic spectrum, i.e. light having a longer wavelength and a lower frequency than visible light. Infrared Spectroscopy generally refers to the analysis of the interaction of a molecule with infrared light.
The IR spectroscopy concept can generally be analyzed in three ways: by measuring reflection, emission, and absorption. The major use of infrared spectroscopy is to determine the functional groups of molecules, relevant to both organic and inorganic chemistry.
1) What is IR Spectroscopy?
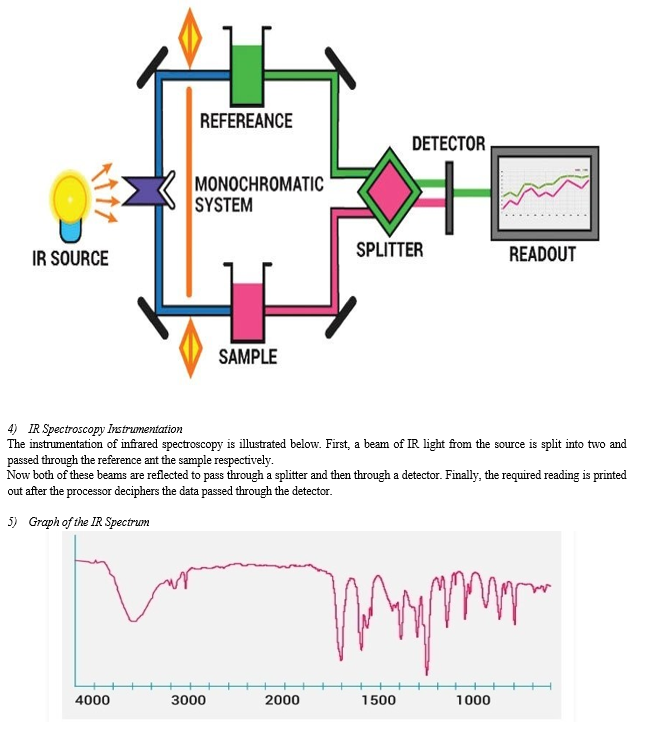
An IR spectrum is essentially a graph plotted with the infrared light absorbed on the Y-axis against. Frequency or wavelength on the X-axis. An illustration highlighting the different regions that light can be classified into is given below.
IR Spectroscopy detects frequencies of infrared light that are absorbed by a molecule. Molecules tend to absorb these specific frequencies fo light since they correspond to the frequency of the vibration of bonds in the molecule.
The energy required to excite the bonds belonging to a molecule, and to make them vibrate with more amplitude, occurs in the infrared region. A bond will only interact with the electromagnetic infrared radiation, however, if it is polar.
The presence of separate areas of partial positive and negative charge in a molecule allows the electric field component of the electromagnetic wave to excite the vibrational energy of the molecule. The change in the vibrational energy leads to another corresponding change in the dipole moment of the given molecule. The intensity of the absorption depends on the polarity of the bond. Symmetrical non-polar bonds in N=N and O=O do not absorb radiation, as they cannot interact with an electric field.
2) Samples in Infrared Spectroscopy
The samples used in IR spectroscopy can be either in the solid, liquid, or gaseous state.
- Solid samples can be prepared by crushing the sample with a mulling agent which has an oily texture. A thin layer of this mull can now be applied on a salt plate to be measured.
- Liquid samples are generally kept between two salt plates and measured since the plates are transparent to IR light. Salt plates can be made up of sodium chloride, calcium fluoride, or even potassium bromide.
- Since the concentration of gaseous samples can be in parts per million, the sample cell must have a relatively long pathlength, i.e. light must travel for a relatively long distance in the sample cell.
Thus, samples of multiple physical states can be used in Infrared Spectroscopy.
3) Principle Of Infrared Spectroscopy
- Infrared Spectroscopy is the analysis of infrared light interacting with a molecule.
- The portion of the infrared region most useful for analysis of organic compounds have a wavelength range from 2,500 to 16,000 nm, with a corresponding frequency range from 1.9*1013 to 1.2*1014 Hz.
- Photon energies associated with this part of the infrared (from 1 to 15 kcal/mole) are not large enough to excite electrons, but may induce vibrational excitation of covalently bonded atoms and groups.
- It is known that in addition to the facile rotation of groups about single bonds, molecules experience a wide variety of vibrational motions, characteristic of their component atoms.
- Consequently, virtually all organic compounds will absorb infrared radiation that corresponds in energy to these vibrations.
- Infrared spectrometers, similar in principle to other spectrometer, permit chemists to obtain absorption spectra of compounds that are a unique reflection of their molecular structure.
- The fundamental measurement obtained in infrared spectroscopy is an infrared spectrum, which is a plot of measured infrared intensity versus wavelength (or frequency) of light.
C. TLC Densitometric Method
Nasr Mohamed A.et al.4 developed TLC Densitometric method for estimation of Indacaterol in bulk drug and capsules.
1) Apparatus
- Camag-Linomat 5 auto sampler (Switzerland) was used for data acquisition. The system is equipped with a deuterium and halogen tungsten lamp as a radiation, while a 100 μL syringe (Hamilton, Bonaduz, Switzerland) was used for sample application, the scanning mode is absorbance mode, the slit dimension is 3 mm × 0.45 mm, the scanning speed is 20 mm s–1.
- Pre-coated TLC plates, silica gel 60 GF254 (20 × 20 cm), (Fluka chemie, Switzerland).
2) General Procedure
Preparation of standard solutions
A standard solution of indacaterol (1mg/ml) was prepared by dissolving 100 mg of the drug powder in 50 ml of methanol and the volume was completed to 100 ml with the methanol. Working solution of (100 μg/ml) was prepared from the stock solution by suitable dilution with methanol.
Chromatographic condition for TLC-densitometric method
Analysis was performed on pre-coated 20 × 10 cm TLC aluminum silica gel 60 GF254 plates. Samples were applied to the plates using Hamilton micro syringe (50-μL). Plates were spotted 1 cm apart from each other and 1 cm apart from the bottom edge. The chromatographic tank was presaturated with the mobile phase for 20 min, then developed by ascending chromatography using (60% Methanol: 30%ethylacetate: 10%water by volume) as a mobile phase. The plates were air dried and the spots were scanned at 260 nm with CAMAG TLC scanner. The produced bands were scanned under deuterium lamp at 260 nm. Concentration of indacaterol in the produced bands was appears as peak areas in the TLC-densitometric chromatograms. The produced peak areas were plotted versus analyte concentrations and the regression equation of the linear relation was computed.
Linearity and construction of calibration graphs
Into a 10 mL volumetric flask, aliquot of standard solution equivalent to (1-6 mg) of indacaterol was transferred and diluted to volume with methanol. So the flasks contain (100-600) μg/mL, 10μL of solution was applied to a TLC plate following the above mentioned specific chromatographic conditions and scanned at 260 nm.
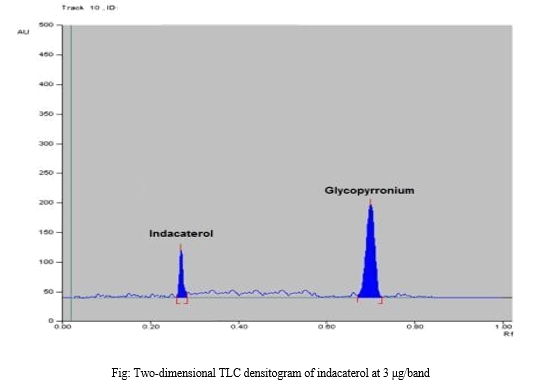
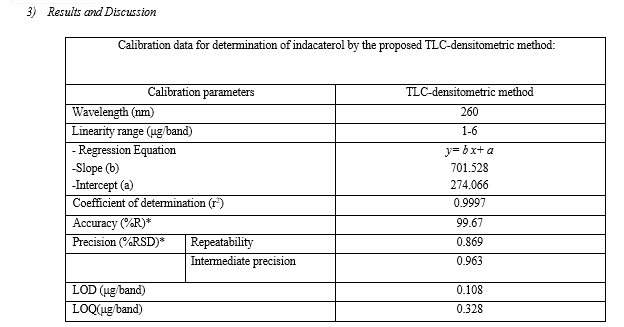
Average of three replicates determinations of three concentrations (1, 2, and 4) μg/band of indacaterol.
4) Conclusion
In this study, TLC-densitometric procedures has been developed and validated for the determination of in pure form and in their pharmaceutical preparation. The developed method is time saving where many bands can be run at the same time. This method is also economic since a small quantity of mobile phase as a developing system was used unlike HPLC procedures. Furthermore, this TLCdensitometric procedure can replace the HPLC method when HPLC requirements are unavailable.
Finally, we can conclude that the described TLC- procedure can be used in routine analysis of indacaterol in pure forms and pharmaceutical dosage form.
D. High Performance Liquid Chromatography method
Y. A. Salem et al.5 Developed high performance Liquid chromatography for determination of Indacaterol adopting Ultraviolet and Fluorescence Detection in pharmaceutical formulation as well as in bulk.
1) Apparatus
Separation was performed with shimadzuTM LC-20A series chromatograph equipped with a 20 μL Rheodyne injector valve and a SPD-20A UV detector operated at 259 nm, and RF-10AXL fluorescence detector operated at (λex/em: 258/421 nm). LC workstation (Nishinokyo- Kuwabaracho, Nakagyo- Ku,Kyoto, Japan), Total Chrom Workstation (Massachusetts, USA) was applied for data collecting and processing. Mobile phase was degassed using Merck solvent L-7612 degasser. A Consort P-901 pH-meter was used for pH measurements.
2) Columns and Mobile Phases
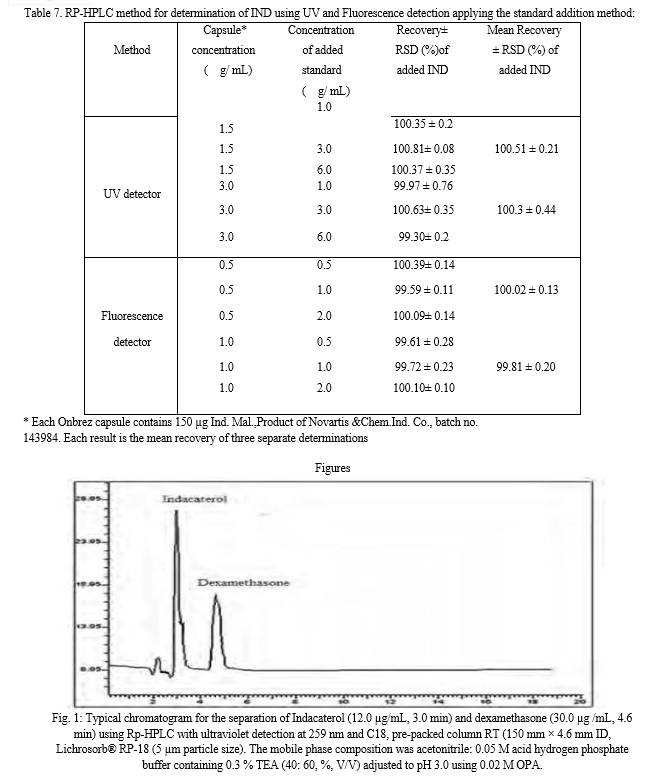
A Hibar® C18, pre-packed column RT (150 mm × 4.6 mm ID, Lichrosorb® RP-18 (5 μm particle size) was used as a stationary phase. The column was operated at ambient temperature. The mobile phase consisted of acetonitrile: 10 Mm acid hydrogen phosphate buffer containing 0.3 % TEA [40: 60]
for both ultraviolet and fluorescence detection. The pH of the mobile phase was adjusted after mixing to 3.0 using 0.02 M OPA and was pumped at flow rate of 1 mL/ min. The mobile phase was shaken on an ultrasonic bath for 5 min and was filtered through a 0.45-μm membrane filter (Millipore,
Ireland).
3) Standard Stock And Working Solutions
Stock solution of 100 μg/mL of Indacaterol (IND) was prepared by dissolving 10.0 mg of Indacaterol (IND) into 100 mL measuring flask containing methanol. The mobile phase was used for further dilution of standard solutions to reach the required concentration range of 2.0-20 μg/mL for ultraviolet detector and 0.05-5.0 μg/mL for a fluorescence detector. The standard solutions were found to be stable for at least one week when kept in the refrigerator.
General procedures and calibration graphs
To a set of 10 mL volumetric flasks, increasing volumes of the standard solution of IND were quantitatively transferred so as to give solutions within the working concentration range of 2-20 and 0.05-5.0 μg/mL for ultraviolet and fluorescence detection, respectively. The flasks were further diluted to 10.0 mL with the mobile phase. Twenty microliter aliquots were injected (in triplicate) at ambient temperature (25ºC) and eluted with the specified mobile phase under the reported chromatographic conditions and the calibration curves were constructed by plotting the peak area ratio [drug/I.S.] against the final concentration of IND (μg/mL). Alternatively, the corresponding regression equations were derived.
Analysis of the studied drug in its capsules
The contents of ten capsules were emptied and mixed well. A weighed quantity of the powder equivalent to 1.5 mg of IND was transferred into a small conical flask and extracted three successive times each with 30 mL of methanol. The extracts were collected, filtered and transferred quantitively into a 100 mL volumetric flask and completed to the volume with the same solvent. All samples were filtered through 0.45 μm sample filters (RC25, Sartorius AG, Gottingen, Germany) prior to injection into the HPLC system. Three different concentrations covering the working concentration range for both detection methods were transferred into two sets of 10 mL volumetric flasks and the procedures for the calibration graph were followed as described under “Construction of calibration curves”.
The nominal content of the capsules was determined using the corresponding regression equation.
4) Results and Discussion
a. Method Development
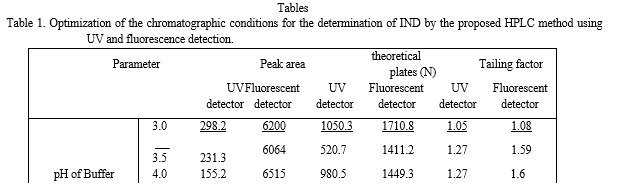
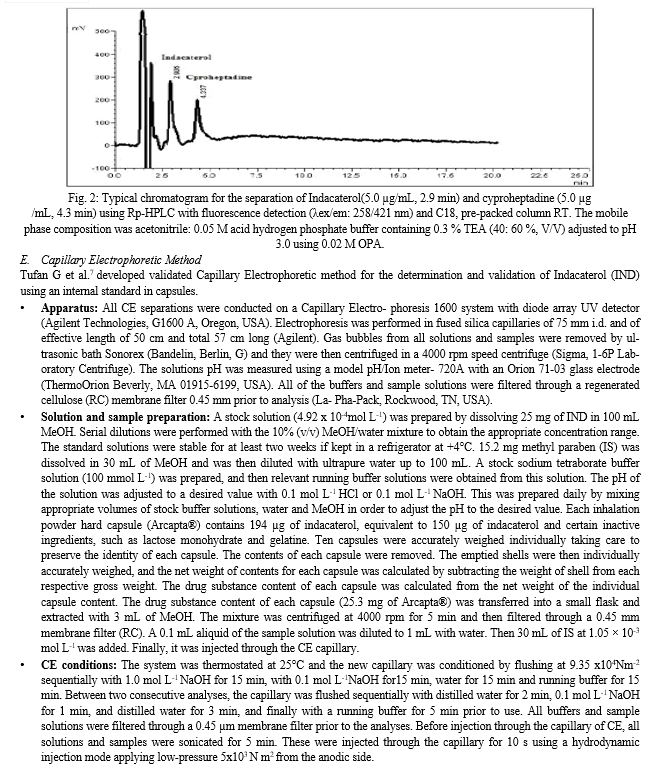
The separation was achieved using a mobile phase of pH 3.0 consisting of acetonitrile: acid hydrogen phosphate buffer containing 0.3 % TEA in a ratio of either [40: 60%, v/v] for both detection modes. Determination of IND within 5 min run time was achieved by the proposed method; figure 1 and 2 represent the obtained chromatogram of IND using ultraviolet and fluorescence detection, respectively.
The most appropriate chromatographic system was developed upon studying the effect of many experimental parameters of the chromatographic system.
-
- The Stationary phase
Two different columns were investigated: Cyano column (250 x 4.6 mm i.d., 5 μm particle size), and Hibar® C18, pre-packed column (150 mm×4.6 mm ID., 5-μm particle size). Experimental studies revealed that the use of C18 column used gives better, symmetrical and well defined peak.
-
- The mobile phase
1.2.a Effect of pH
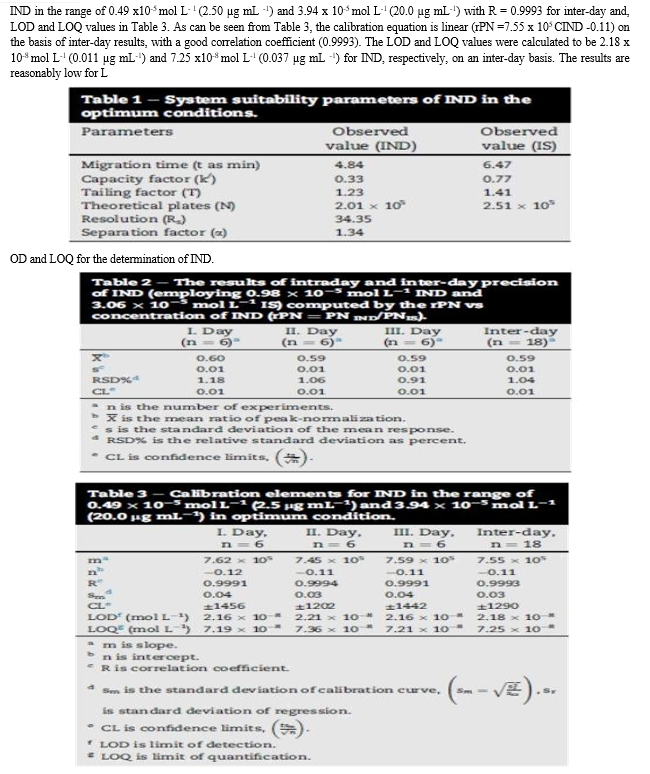
The mobile phase pH was altered using increasing volumes of 0.3 % triethylamine (TEA) over a specified range of 3.0 to 6.0. The retention time of IND was not changed after increasing the pH value up to pH 6.0 as IND will be completely ionized over the investigated pH range (pKa value of IND = 8.5 and 9.7). At pH 6.0, the efficiency of the peak was not suitable for measurements as indicated by the values of N, and tailing factor. For both detectors, a pH value of 3.0 was found to be optimum as it offers resolving and quantitation of both IND drug, and the internal standard in a short run coupled with a good selectivity, peaks efficiency and peak symmetry as presented in table 1.
1.2.b Ratio of Mobile Phase Components
As shown in table 1, increasing ratios of ACN: 0.05 M acid hydrogen phosphate buffer were investigated over the range from 30: 70 to 70: 30. It was found that increasing ratio of buffer to acetonitrile (70:30, v/v) retained IND on the column for more than 8 min. Meanwhile, upon increasing acetonitrile content versus acid hydrogen phosphate (70:30, v/v), it was noticed that the drug was unretained as the retention time was greatly decreased and the drug peak was overlapped with solvent front.
The sensitivity parameter based on the peak area of the drug and the peak efficiency based on number of theoretical plates (N) were the basis of choosing the optimum acetonitrile ratio in the mobile phase.
Thus, the optimum ratio was found to be (40: 60%, v/v) acetonitrile: acid hydrogen phosphate buffer. It provides the best sensitivity, resolution and reasonable run time for both detectors.
1.2.c Type of organic modifier
Different organic modifiers including methanol and n-propanol were investigated as alternates for acetonitrile. Replacement of acetonitrile with methanol retained IND on the column for more than 8 min with lower sensitivity. Also, the replacement of phosphate buffer with water resulted in decreased peak efficiency as revealed by lower number of theoretical plates, and broadening of IND peak. Thus, acetonitrile and sodium phosphate were the ideal selection for the chromatographic separation.
1.2.d The Flow rate
The effect of flow rate was studied to optimize the chromatographic efficiency of the proposed method and improve the resolution of the eluted peaks. The flow rate was changed over the range of
0.2-1.2 mL/min and a flow rate of 1 mL/min was the optimum for good separation in a reasonable time adopting both detectors. The results are shown in Table 1.
1.2.e The Choice of the Internal Standard:
The use of internal standard is very important for providing a well-developed accurate and precise HPLC method. Different drugs such as dexamethasone, rosuvastatin, ezetimibe, cyproheptadine, terbutaline, and ciprofloxacin were investigated as possible internal standards. Dexamethasone and cyproheptadine were the best internal standard that provides excellent separation of its peak from the intact drug for both UV and fluorescent detectors, respectively. While, other drugs under investigations gave overlapping peaks with the drug. The used concentrations of dexamethasone and cyproheptadine are 30.0 and 5.0 μg/mL for ultraviolet and fluorescence detection, respectively.
Difference in their elution order was revealed by the difference in lipophilicity between IND and the internal standard as indicated by Log P values which are 3.3, 1.93 and 5.027 for IND, dexamethasone and cyproheptadine, respectively as shown in fig.1 & 2.
b. Method Validation
The developed analytical method was then subjected to method validation according to ICH Q2 (R1) guidelines8. The following parameters were considered: linearity, sensitivity, LOD, LOQ, accuracy and precision.
2.1 Linearity
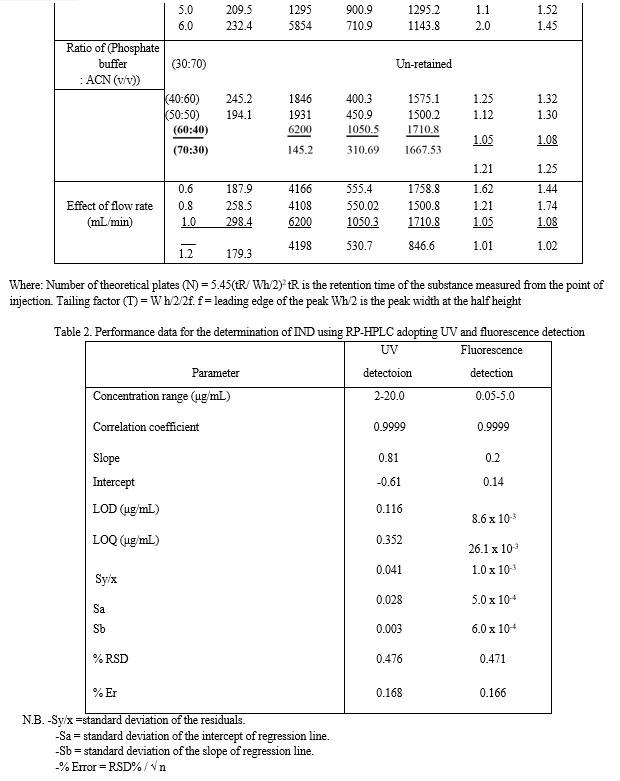
Under the above described experimental conditions, a linear relationship was established by plotting the peak area ratio [drug/I.S.] against the drug concentration in μg/mL. The concentration ranges were found to be 2.0-20 μg/mL and 0.05-5.0 μg/mL for ultraviolet and fluorescence detection, respectively (Table 2).
Linear regression analysis of the data by the proposed method gave the following equations:
PA = -0.61 + 0.81 C (r =0.9999) ultraviolet detector PA =
0.14 + 0.2 C (r =0.9999) fluorescence detector
Where, PA is the Peak area ratio, C is the concentration of the drug (μg/mL) and r is correlation coefficient. Statistical analysis of the data gave high value of the correlation coefficients (r) of the regression equations, small values of the standard deviation of residuals (Sy/x), intercepts (Sa), and slopes (Sb), and small values of the percentage relative standard deviations and the percentage relative errors (Tables 2). These data proved the linearity of the calibration graph.
2.2 Limit of Quantification (LOQ) and Limit of Detection (LOD)
LOQ and LOD were calculated according to ICH Q2 (R1) recommendations6 using the following equations:
LOQ = 10 Sa /b and LOD = 3.3 Sa /b
Where, Sa = standard deviation of the intercept and b = slope of the calibration curve
The limit of quantitation (LOQ) was determined by establishing the lowest concentration of the analyte that can be measured and below which the calibration graph is non linear. While, the limit of detection (LOD) was determined by establishing the minimum level at which the analyte can be reliably detected. Values of LOQ were 0.352 and 0.026 μg/mL, whereas, values of LOD were 0.116 and 0.009 μg/mL using UV and fluorescence detection, respectively.
2.3 Precision
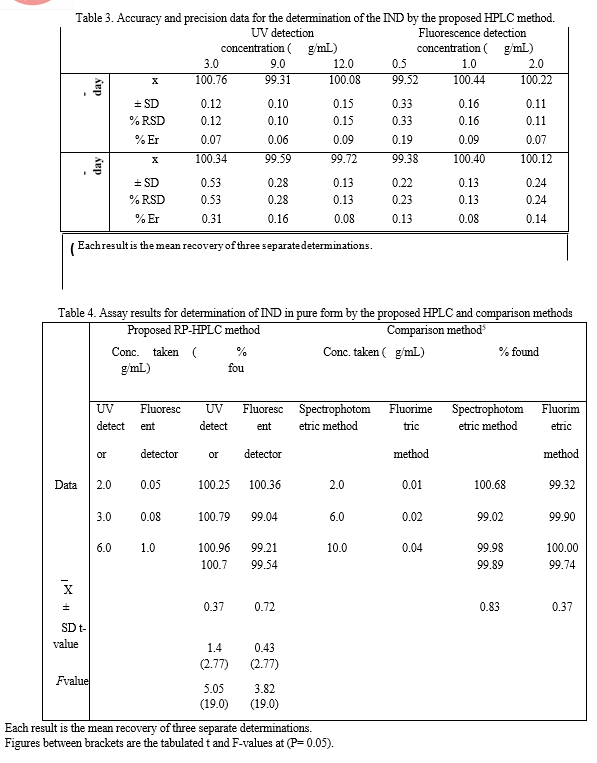
The intra-day and inter-day precisions of the proposed HPLC method was examined by triplicate analysis of IND at three different concentrations 3.0, 9.0 and 12.0 μg/mL and 0.5, 1.0 and 2.0 μg/mL for ultraviolet and fluorescence detection, respectively at one day and for three consecutive days. The precision of the proposed method was satisfactory, as indicated by the low values of SD and RSD, also the low values of % Er indicates good accuracy of the method (Table 3).
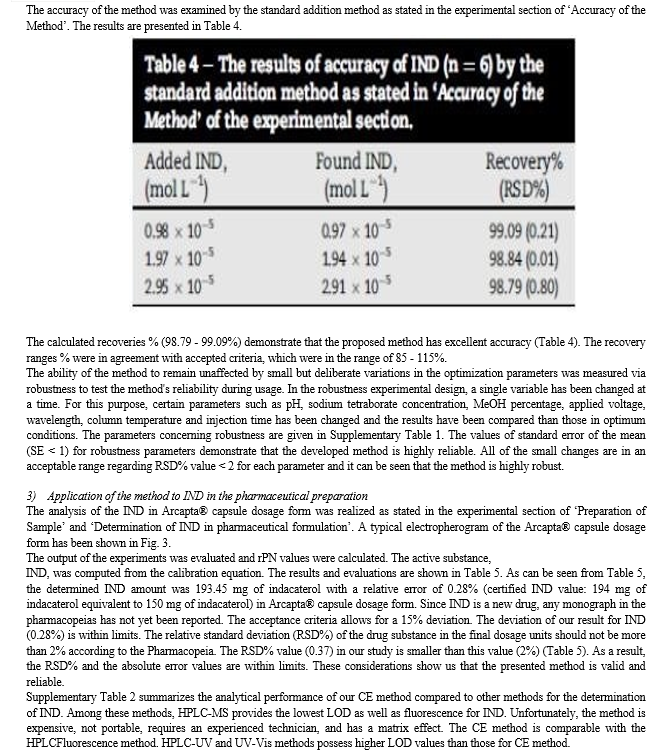
2.4 Accuracy
To prove the accuracy of the proposed method, the results of the assay of the studied drug in pure and dosage form were compared with those of the comparison method.
Statistical analysis of the results obtained by the proposed and the comparison method using Student's t-test and variance ratio F-test revealed no significant difference between the performance of the two methods regarding the accuracy and precision, respectively (Tables 4 and 5) ,since the calculated values did not exceed the tabulated ones.
2.5 Robustness
The steadiness of the peak area of IND with the intentional minor changes in the chromatographic conditions approve the robustness of the proposed method; these changes include; pH (3.0 ± 0.1), proportion of mobile phase (Buffer: ACN, 40: 60 v/v ± 2%), and flow rate (1.0 ± 0.1) mL/min for both detection modes. The peak area of IND drug was not highly influenced by these deliberate minor changes (Table 6).
2.6 Selectivity
The selectivity of the proposed method was proven by its ability to determine IND in its capsules without interference from the common excipients. The results were summarized in Table 5.
2.7 System suitability test (SST)
Evaluation of SST parameters was performed during the development and optimization of the method (Table 1). Moreover, to ascertain the effectiveness of the final operating system it was subjected to suitability testing. The test was performed by injecting the standard mixture in triplicate and the parameters were calculated as reported by the USP. SST parameters include tailing factor (T) and column efficiency (number of theoretical plates, N).
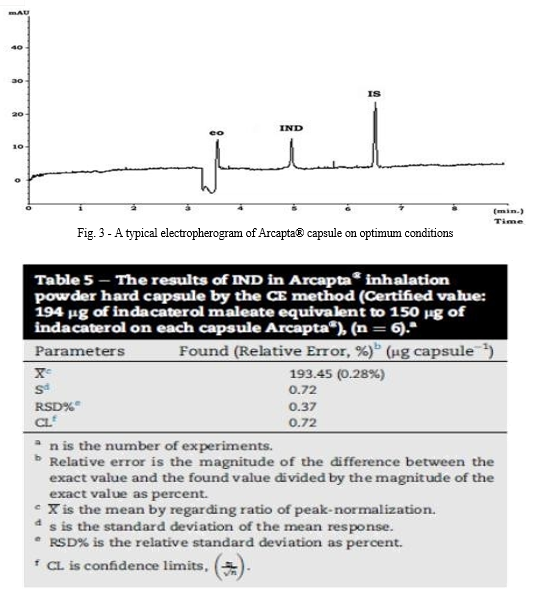
c. Assay of Pharmaceutical Preparation
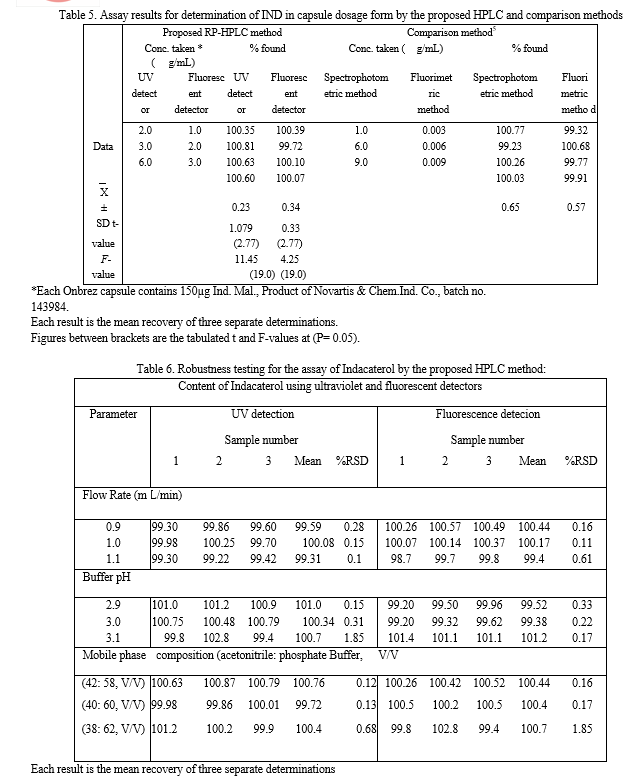
The proposed methods were applied successfully to the determination of IND in its capsules where no apparent interference from the capsule excipients (lactose) was noticed as illustrated by the placebo peaks. The potency of very low dose of Indacaterol (150 μg/mL) in its onbrez capsule combined by fast onset of action together with a dosing regimen compatible with once-daily dosing .Standard addition method was applied to test the validity of the proposed methods. The recovery of IND, was determined by adding a known amount of pure drug at three different concentrations of 1.0, 3.0, and 6.0 μg/Ml for UV detector and 0.5, 1.0, and 2.0 μg/mL for fluorescence detector to previously analyzed capsule solution at two different concentrations 1.5 and 3.0 μg/mL for UV detector and 0.5, and 1.0 μg/mL for fluorescence detector. These concentrations of the pure drug were added in separate flasks to each capsule concentration and each solution was reanalyzed for the total drug content. The analysis was carried out in triplicate and was performed as described under “Construction of calibration curves”. The obtained results are shown in table 7. These control experiments eliminate the interference due to interactions of other constituents encountered in the system or caused by the bulk production.
5) Conclusion
The proposed method applies a validated HPLC to determine IND in pure form and in its capsule dosage form. The proposed method presents rapid (retention time is less than 5 min), sensitive (LOD values were 0.116 and 0.004 μg/mL adopting ultraviolet and fluorescence detection, respectively.
Moreover, the proposed method was applied efficiently to analyze dosage form capsules in quality control laboratories.


Conclusion
A rapid, accurate, selective, reliable and environmentally friendly new capillary electrophoretic method for the determination of IND was developed and is validated in this study. The method is simple, easy to apply and IND was determined within 8 min. The method is also cheap and has many advantages over conventional chromatographic techniques, including reduction in the use of organic solvents, small sample volume, and increased efficiency and resolution. To the best of our knowledge, this is the first CE study concerning the determination of IND. The method has been validated with respect to precision, linearity range and accuracy, LOD, LOQ, specificity and robustness. All the system suitability parameters gave good results. The proposed method has been successfully applied to the analysis of IND in the pharmaceutical preparation of Arcapta® capsule dosage form.
References
[1] https://go.drugbank.com/drugs/DB05039 [2] https://pubchem.ncbi.nlm.nih.gov/compound/Indacaterol [3] Salem YA, Sherbiny DT, Wasseef DR and Ashry SM. Spectroscopic study on Indacaterol maleateAnalytical applications for quality control of capsules. International Journal of Pharma Sciences and Research. 2015; 6(3): 592-605. [4] Nasr MA, El-Abasawy, Khalid AS, Ahmed AA, Ahmed O, Ayman OE. Application of TLC Densitometric and UV Spectrophotometric techniques for simultaneous determination of Indacaterol and Glycopyrronium in Inhalation Capsule used for COPD. European Journal of Biomedical and Pharmaceutical Sciences. 2018; 5: 80-89. [5] Salem YA, Sherbiny DT, Wasseef DR and Ashry SM. HPLC Determination of Indacaterol Maleate in Pharmaceutical Preparations Adopting Ultraviolet and Fluorescence Detection. International Journal of Pharma Sciences and Research. 2015; 6(2): 1324-1332. [6] https://www.ema.europa.eu/en/documents/scientific-guideline/ich-q-2-r1-validation-analyticalhttps://www.ema.europa.eu/en/documents/scientific-guideline/ich-q-2-r1-validation-analytical-procedures-text-methodology-step-5_en.pdfprocedures-text-methodology-step-5_en.pdf [7] Tufan G, Muzaffer T, Ulku DU. A validated capillary electrophoretic method for the determination of indacaterol and its application to a pharmaceutical preparation. Journal of Food and Drug Analysis. 2018; 26: 842-848. [8] Emotte, C., Heudi, O., Deglave, F., Bonevie, A., Masson, L., Picard, F., & Kretz, O.. Validation of an on-line solid-phase extraction method coupled to liquid chromatography-tandem mass spectrometry detection for the determination of indacaterol in human serum. Journal of chromatography B,2012;895:1-9. [9] Ammari, W.G., Al-Qadhi, Z., Khalil, M., Tayyem, R., Qammaz, S., Oriquat, G., & Chrystyn, H.. Indacaterol determination in human urine: Validation of a Liquid-Liquid Extraction and Liquid Chromatography-Tandem Mass Spectrometry Analytical Method. Journal of aerosol medicine and pulmonary drug delivery, 2014; 27: 1-9. [10] El-Ashry, S. M., EL-Wasseef, D. R., El-Sherbiny, D. T., & Salem, Y. A.. Spectrophotometric andSpectroflurimetric Determination of indacaterol maleate in pure form and pharmaceutical preparations: Application to content uniformity. Lumintscence, 2015, DOI: 10.1002/bio.2838.
Copyright
Copyright © 2022 Sachin Laxman Ingale , Dr. Hingane L. D., Prof. Nakhate S. T. . This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.

Download Paper
Paper Id : IJRASET44995
Publish Date : 2022-06-28
ISSN : 2321-9653
Publisher Name : IJRASET
DOI Link : Click Here
 Submit Paper Online
Submit Paper Online