

Ijraset Journal For Research in Applied Science and Engineering Technology
New-Generation Drug Discovery using Machine Learning
Authors: Niharika Gupta, Priya Khobragade
DOI Link: https://doi.org/10.22214/ijraset.2023.52112
Certificate: View Certificate
Abstract
Finding innovative molecules with specific chemical properties to treat diseases is one of the goals of drug discovery. Recent years have seen the production of a sizable volume of biological data from many sources. These statistics and molecular analyses have been used to determine the most effective medications. Medical research often frustrates people and is far more expensive. The work at hand is made easier by having the ability to predict whether a medicine will be active or not. The information about the drug can also be used to develop other drugs. One application that makes use of machine learning to enhance decision-making in pharmaceutical data across numerous applications is quantitative structure activity relationship (QSAR) analysis. Machine learning-based predictive models have recently gained a lot of attention in areas outside of preclinical research. Costs and research times associated with finding new drugs are considerably decreased at this stage. Drug research is growing and more commonly utilising machine learning, algorithms for pattern recognition, knowledge of mathematical correlations, and knowledge of the chemical and biological characteristics of molecules. The necessity for a sizable volume of data, the incapacity to interpret the data, and other issues are further restrictions. Without the need for computational resources, massive amounts of data can be analysed using both physical models and machine learning approaches.
Introduction
I. INTRODUCTION
The drug discovery process aims to find effective molecules for illness detection and therapy. Precision medicine considers a person's unique genetic makeup and surroundings for disease treatment and prevention. This approach helps medical professionals anticipate which strategies will be helpful for specific demographics. Developing a new pharmaceutical takes 10 to 15 years of research and testing. Investigating chemical compounds can simplify treatment development. Machine learning techniques aim to reduce the cost of drug discovery research. The ChEMBL database provides details on chemical and biological properties of substances. This study focuses on acetylcholinesterase. Key terminologies will be clarified.
- Drug Development: The identification and confirmation of a disease target and the development and synthesis of a chemical molecule to interact with that target can be used to summarise the drug discovery process. The initial phase in the medication development process is the identification of a condition with well defined symptoms that lowers quality of life. A chemical (which could be a simple molecule or a complex protein) or chemical combination that lessens symptoms without significantly harming the patient is frequently viewed as a desired medication.
- Bioinformatics: The exponential growth of biological data is the main focus of the area of bioinformatics, It prompted the creation of both primary and secondary databases on the structures and sequencing of proteins and nucleic acids. It is a field of study that makes use of computer techniques, statistical instruments, algorithms, and mathematical ideas to study biological data. Bioinformatics is thought to be crucial for understanding the intricate cell mechanisms. Bioinformatics is also found to be extremely useful for assessing clinical samples by biomedical researchers.
- Artificial intelligence (AI): The study of artificial intelligence examines the many organisational structures and applications of different data-processing systems. The term "Computational Intelligence" is a catch-all term that encompasses a broad range of fields, including machine learning, fuzzy models, neural networks, probability theory, statistics, and pattern recognition. These and other domains have a lot in common with the idea of AI. It can solve a variety of problems relating to human intelligence and, consequently, the computer representation of these processes. From theoretical understanding to practical data, artificial intelligence research has now advanced.
- Machine Learning(ML): Machine learning, a branch of artificial intelligence, is based on computational and mathematical theory. The foundation of machine learning is the development of models through exposure to training data. The usage of ML algorithms in the pharmaceutical industry has grown dramatically during the last two years.
Instead of being limited to particular data kinds in the past, such as protein sequences and chemicals, it may now be used with a variety of data types and techniques, such as imaging and protein structures. Gradually more machine learning is being used in the drug development process, and it is producing effective results by using algorithms for pattern recognition, clever mathematical correlations, etc.
5. Deep Learning: A machine learning component called "deep learning" can extract characteristics with a higher level of detail from multiple layers of input data. A vast field that is currently highly valued is deep learning. Deep learning algorithms are now more widely applied in corporate settings and across a range of scientific fields. But how exactly does deep learning work? Deep learning is based on neural networks that often have many layers and allow for data changes between them. Its continued widespread use is the consequence of sincere and careful innovation. Therefore, deep learning models can be created using a method known as greedy layer-by-layer.
II. METHODOLOGY
Even if the experimental design process is shared by all study areas in some ways, ML tactics need to be cross-disciplinary. The ML technique's steps that are unique to drug discovery are as follows: The key five steps are data collection, mathematical descriptor creation, best variable selection, model training, and model validation.
- Data Collection: Getting the data set is the initial stage, and it must meet certain requirements. It must have qualities that are simple to manufacture and manage in the lab in addition to physical-chemical properties that support absorption, selectivity, and low toxicity. This is due to the fact that large proteins or complex chemicals are not frequently used in the pharmaceutical industry. It typically interacts with peptides and small molecules as the main chemical kinds. SMILES (simple molecular-input line-entry system) and To facilitate handling and study of these compounds, the sequencing and structure of small molecules and peptides are represented in FASTA formats. DrugBank, PubChem, ChEMBL, and ZINC are just a few of the many public data repositories currently available in the field of drug development.
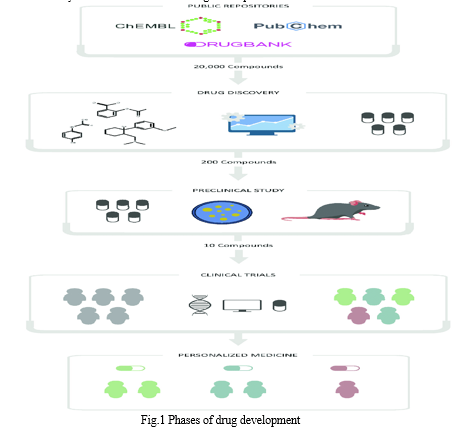
2. Creation of mathematical descriptors: While some machine learning (ML) models do not require labeling, supervised learning models are often used in the field of drug development. In this case, an accurate determination by scientists will greatly improve the experimental process. The generation of mathematical descriptors provides a data set that the ML model can process. This data set is divided into two subsets, one of which (Fig.2) contains most of the data used to train the model, and the second (Fig. 3) contains a smaller part of the data used to test the model. The training set is searched for the best selection of variables with the required and accurate data. However, supervised learning models are commonly used in drug discovery, although some machine learning (ML) models do not require labeling. In this case, the precise labeling used by the researchers will greatly improve the experimental workflow. The development of the mathematical descriptors results in a set of data that the ML model is able to process. This dataset is divided into two subsets, the first of which (Fig. 2) has more of the data used to train the model than the second (Fig. 3), which contains more of the data used to test the model. In the training set, the best variables with the necessary and precise data are sought after.
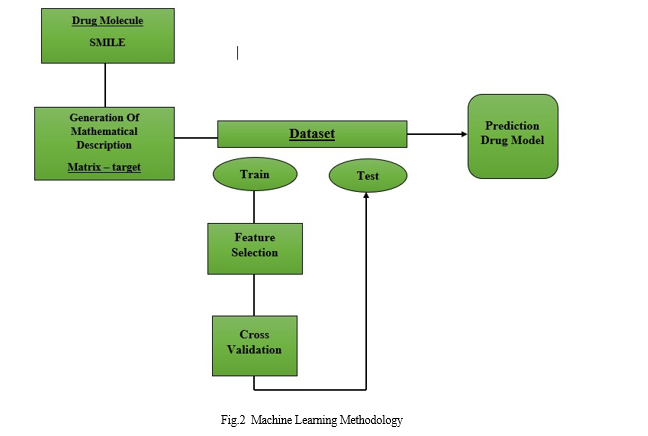
3. Finding the best set of variables First: FS methods are used to select a subset of the initial set of features, but the content of the variables is ignored. You must consider the algorithms and their input parameters. These should be chosen carefully to ensure they are appropriate for the task at hand and the amount and type of data available. Because it provides a scientifically understandable rationale, most researchers use these techniques when developing experimental designs.
4. Model Training: The model is trained after the ideal set of variables has been identified.The experiment is then repeated using practical data. To ensure that the model can be used with unknown inputs, overtraining should be avoided. In these cases, cross-validation (CV) techniques are often used. With CV, you can assess performance, predict performance using dummy data, and monitor the generalization of your model during training.
5. Validation of the model: The initial data set is divided again into three groups for each sample.The training set and the validation set are two subsets. Figure 2 shows the evolution of the CV strategy over 10 iterations. The blue set serves as the training set and the red set as the validation set for each of these runs. As a result of the CV process, the optimal parameter combinations are selected for each approach. These criteria are used to evaluate the performance of each model.Finally, after retrieving a test set extracted from the original set, the best model built using the CV method was demonstrated (Fig. 2). A new predictive drug model may have been created if the validation results are statistically significant.
Many industries use machine learning techniques and in particular, more research has been published recently. There are few machine learning related articles on open access platforms dedicated to drug manufacturing.
III. DATABASES, SOFTWARES, PACKAGES and THEIR REPRESENTATION
A. Databases
ChEMBL:- The ChEMBL database data was compiled manually based on literary works. The European Bioinformatics Institute and the European Molecular Biology Laboratory (EMBL) made this database available in 2002. This database contains over 1.9 million chemical compounds, was last updated in 2018 and is still growing.According to ChEMBL, these connections span over 10,000 drugs and over 12,000 targets. Since it's a live dataset, you can access it by integrating it with the API and pulling the data from there.
- DrugBank: DrugBank is one of the most popular databases and most widely used source of drug information. The first version of this database dates back to 2006. This bioinformatics and chemoinformatics database contains full drug targeting and extended drug data.DTI DrugBank's first partner sources were books, journals and other electronic databases. In addition, all data can be downloaded free of charge.
- PubChem: Data on chemical compounds and related biological activities are stored in PubChem. Sub-databases on substances, compounds and bioassays make up this database. The primary storage location for the chemical data provided by each data source is the substance.unique chemical structures transferred from the substance database to the compound database. The BioAssay database contains all biologically relevant information on this chemical compound.
B. Softwares
Given the current interest in deep learning applications, various software tools have been developed to facilitate pattern interpretation. Most of the function assignment algorithms presented in this article are based on Captum, a module in PyTorch's deep learning and machine differentiation suite. A popular package called Alibi provides instance-specific justifications for certain models built with the TensorFlow or scikit-learn libraries. Some of the explanatory strategies used in are anchor, descriptive, and counterfactual explanations.
C. Packages
Based on previous work, Sakakibara created the Comprehensive Predictor of Interactions between Chemical Compounds and Target Proteins website, which uses SVM as a predictor of drug-target interactions (DTI). It seems that this server is no longer available. To combine chemoinformatics, bioinformatics, proteochemistry, and chemogenomics to predict DTI, Cao developed the random forest-based PyDPI tool. The proposed method requires selection of chemical properties and usesready-made vocabularies for categorization.This package can be used to create web servers and provides an interface to databases such as PubChem, Drugbank, Uniprot, and the Kyoto Encyclopedia of Genes and Genomes (KEGG). PreDPI-Ki, a web-based service, was developed by the same team in the same year. PreDPI-Ki is based on a random forest predictor and takes into account the binding affinities of DT pairs to better predict interactions.
D. Representation
- SMILE Code: Text-encoding a molecule's structural data is the best method for representing it. This process converts graphical structural data into text for use in the machine learning pipeline.The SMILES (Simplified Molecular -Input Line Entry System) depiction is the one that is used the most frequently. Once the conversion is finished, we can process the medication and employ various algorithms like NPL to predict its characteristics, chemical interactions, and side effects.
- Molecular Fingerprint: One of the many ways a medicine might be represented in the machine learning input pipeline is by its chemical fingerprint. The most frequent format is binary digits, which can reveal whether or not a molecule contains a particular substructure.
It is obvious that employing a molecule as an informational vector is a process that cannot be reversed. Because the fingerprint cannot be extracted from the molecule, this technique causes information loss.
3. FASTA Code: The text-based FASTA format, which is used to represent either nucleotide sequences or peptide sequences, uses single-letter codes to represent base pairs or amino acids. A sequence in the FASTA format is composed of a single line of description and multiple lines of sequence data. The description line is distinguished from the sequence data by the greater-than (">") character in the first column. All text lines should be no more than 80 characters, it is recommended.
IV. ALGORITHMS
- Naïve Bayes:- This method of drug discovery has been used to anticipate possible drug targets. They particularly developed a Bayesian model that integrates numerous data sources, such as information on known adverse effects or gene expression, and they were able to produce a model that was 90% accurate for more than 2,000 chemicals. They also developed the screening method's experimental validation. They create two descriptors using ChEMBL data and confirm their predictions using docking techniques. Then, for the treatment of HCV, they forecast drugs with an AUC of 80% that are multi-target. Using interactions with four distinct groups of proteins (enzymes, ion channels, GPCRs, and nuclear receptors), they developed a model for the prediction of ligand-target interactions with a 95% accuracy.
- Support Vector Machine(SVM):- SVM plays a vital role in drug development because of its ability to distinguish between active and inactive compounds as well as its capacity to train the regression model. To understand how medicines and ligands interact, regression models are essential. Additionally, it has the ability to deal with difficult problems that are complex, non-linear, high-dimensional, and noisy. They have been used to classify drugs with an accuracy value of 83.9% based on their classification in KEGG. a new method that predicts intricate drug-target interaction networks from interaction matrices with an F1 value of 80% accuracy. It is also possible to predict the stability in human liver microsomes with values close to 70% accuracy of validation by computing several molecular descriptors and chemical indices from 25 ChEMBL datasets.
- Random Forest(RF):- A collection of decision trees is called Random Forest. We'll find a range of desirable trees in the random forest. If we need to categorise an item based on an attribute, each tree will vote for a class, and the decision forest will choose the class with the most votes. A single decision tree typically does not produce high-performance results. To reduce large variance, the tree is frequently trimmed using cross validation or model complexity parameters. It has been demonstrated that employing RF models improves the performance of several Decision Trees under LBVS. The benefits of Random Forest enhance the prediction of QSAR data. High prediction accuracy and descriptor selection are inherent characteristics. Regardless of the issue at hand, this is one of the most often used machine learning techniques. RF is without a doubt among the top models in terms of performance, speed, and generalizability, even though it is impossible to choose one model as the best for all issue kinds. They were able to predict compound-protein interactions with an accuracy of more than 90% using 211,888 compound-protein interactions from BindingDB in an mRMR (max relevance and min redundancy) dimensionality reduction scheme, the descriptors produced by Open Babel, and the enrichment scores of each protein from GO and KEGG. There has been a new proposal for a tree-based model with AUC values greater than 90%. It is determined with RDKit using the molecular descriptors. In this model, features are extracted using the Relief method, and the classifier is determined using Graph Based Semi-supervised Learning.
- Artificial Neural Network(ANN):- The input layer of an ANN is made up of the neurons known as input nodes, which receive information from the outside environment. Additionally, the network requires output nodes that broadcast ANN findings and are positioned in the hidden layer. The rest of the nodes, known as hidden nodes, are organised into one or more hidden layers and are in charge of relaying information among the network's neurons. They employed 1D descriptors to describe the general structure of each compound (molecular weight, hydrogen bond count, etc.), and 2D descriptors to describe the presence or absence of functional groups inside the molecule. An ANN with both sorts of descriptors had an accuracy of 89% and produced the best results. They build six distinct types of descriptors with a deep learning model, with an accuracy of 86%, to predict the early carcinogenesis of drugs that are suggested to be medicinal using 1003 chemicals from the Carinogenic Potency Database.
- K Nearest Neighbor:- Can be used to solve classification and regression problems. Its main use is in classification problems. Before a new instance is ranked based on the majority of its k neighbors, KNN stores all previously ranked cases. To determine which of its k nearest neighbors occurs most frequently, the distance function is used.These measurement distances include Makowski, Hamming, Manhattan, and Euclid distances to name a few. When using ANN modeling, K selection can be difficult. The new chemical is predicted from k neighbors, although this method is prone to noisy data. In order for the molecule to make accurate predictions, the formation data must be accurately classified.
- Decision Tree:- Decision trees, a supervised learning technique, are commonly used in classification problems. Decision trees are used to classify data and make recommendations based on a set of decision rules. In the pharmaceutical industry, decision trees are used to solve a variety of problems such as: B. compound profiling, combinatorial library development, prediction of drug similarity, etc. Decision trees are also used to evaluate ADME properties, metabolic stability, drug penetration, permeability, distribution and solubility of the p-glycoprotein ADME to predict. Decision tree models are simple and easy to master, comprehend and understand.
- Lazy Regressor:- One such device is Lazy Regressor, which evaluates the system studying fashions which might be maximum possibly to be passable. Use Lazy Classifier and Lazy Regressor Lazy Predict to predict binary and continuous variables respectively. This is the best way to efficiently train a model as it simplifies the training process and allows you to train 30 models at once.
V. CHALLENGES
- The challenges of creating precise Drug Target Identification projections can be separated into two categories: database-related challenges and computation-related challenges. Using a variety of prediction techniques, one can typically overcome computational obstacles depending on the work at hand. However, there are substantial issues with the databases' origins.
- The majority of research carried out by pharmaceutical corporations relies on the integration of heterogeneous data, which has distinct difficulties when used in contexts and dimensions ranging from large molecules to singular persons. For managing several sources, a high level of artificial intelligence must be attained, and it must be bolstered with a greater comprehension of the data gathered. In order to merge the various types of data, contemporary data connectors are suggested. Finally, these data interfaces assist in assigning original data.
- Because diverse classification algorithms involve ambiguous decision-making, the accessibility issue adds another challenge to medication discovery. To assess the outcomes of drug development, many pathways must be known. The ability to discover possible therapeutic targets is thereby improved, and the confidence in interpretability must increase due to the large number of assembled qualities. Drug development can make use of a variety of techniques, to understand and interpret the outcomes, such as SVM, Machine Learning, Random Forest, and deep learning algorithms. In order to foster interpretability, it is therefore more beneficial in finding novel medication targets and many assembled aspects.
VI. FUTURE SCOPE
-
Future research need to awareness on strategies that employ plenty of similarities. Techniques that exclusively use one type of similarity are less likely to produce accurate findings than ensemble-based models. For example, repurposed drugs have been located via chance, pharmacological research, or retrospective scientific study (in conjunction with reading side effects). Research is now concentrating on the most effective methods to adopt a more comprehensive, systemic approach in light of the early examples' surprisingly successful repurposings (using thalidomide for morning sickness instead of multiple myeloma, sildenafil for angina instead of erectile dysfunction, and minoxidil for hair loss).
In order to enhance the ability of deep learning algorithms to predict biomarkers, adverse effects of medications, and therapeutic outcomes, medical science and online innovation have been integrated. Using specific software, clinical trials can be successful. In order to encourage possible investments in pharmaceutical companies, this is done. Plans for drug discovery and development in the future call for addressing every aspect with AI technologies. For new applications, AI needs to coordinate theoretical outcomes such as chemical data, omics data, and medical data. We also anticipate that further approvals will be required for drug discovery campaigns.
???????VII. ???????ACKNOWLEDGEMENT
The author thanks Prof. Priya Khobragade for her expert advice and ongoing support throughout the study.They also thank Prof. Minakshee Chandankhede for his careful supervision of the improvisation. .
Conclusion
Machine learning models can take the role of more traditional approaches like PPT inhibitors and macrocycles in the realm of medicine by making predictions based on learnt data inside of a preset framework, i.e., the compound structure. Deep learning models can also incorporate chemical structures and QSAR models from pharmaceutical data because they were pertinent for compounds with the right attributes and had a high clinical trial success rate. Deep learning techniques and machine learning algorithms are frequently employed in the pharmaceutical sector. The use of ML algorithms has helped solve a number of issues in drug development and healthcare service hubs, particularly with regard to image analysis and omics data. AI technology has improved by foraying into the realm of computer-aided drug discovery in an effort to recover its once-powerful data mining capabilities. The growth of data will be beneficial for machine learning techniques and fields. The application of these models in cheminformatics, and more specifically in drug discovery, has greatly benefited the pharmaceutical industry. The use of descriptors derived from the structure of peptides or small molecules was the sole method accessible up until this time. Recently, graph-based molecules have been directly recreated using ANN. Researchers are searching for novel medications, therapies, or cures that are more effective than those that are now available as the field matures. The development of novel, intensely targeted treatment approaches that will ultimately improve patients\\\' health and quality of life depends on an understanding of the underlying mechanisms of disease progression, the side effects of already prescribed medications, and the genetic make-up of the individuals.
References
[1] Speck-Planche, M.N. Cordeiro, “Computer-aided drug design,synthesis and evaluation of new anti-cancer drugs”, Curr Top MedChem. [Epub ahead of print], 2013. [2] “Drug discovery hit to lead”, Available from in Wikipedia.org/wiki/Drug_discovery_hit_to_lead, cited march 26 2012. [3] Senior, A. W. et al. Improved protein structure prediction using potentials from deep learning. Nature 577, 706–710 (2020). [4] Schneider, P. et al. Rethinking drug design in the artifcial intelligence era.Nat. Rev. Drug Discov. 19, 353–364 (2020). [5] Karpov, P., Godin, G. & Tetko, I. V. Transformer-CNN: Swiss knife for QSAR modeling and interpretation. J. Cheminform. 12, 17 (2020). [6] Zhang, R., Li, C., Zhang, J., Chen, C. & Wilson, A. G. Cyclical stochastic gradient MCMC for Bayesian deep learning. Preprint at https://arxiv.org/abs/1902.03932 (2019). [7] Scalia, G., Grambow, C. A., Pernici, B., Li, Y.-P. & Green, W. H. Evaluating scalable uncertainty estimation methods for deep learning-based molecular property prediction. J. Chem. Inf. Model. 60, 2697–2717 (2020). [8] Rifaioglu AS, Atas H, Martin MJ, et al. Recent applications of deep learning and machine intelligence on in silico drug discovery: methods, tools and databases. Brief Bioinform,2018. [9] Patel L, Shukla T, Huang X, Ussery DW, Wang S. Machine Learning Methods in Drug Discovery. Molecules 2020. [10] Lavecchia A. Machine-learning approaches in drug discovery: methods and applications. Drug discovery today 2015. [11] Tong WD, et al. Decision forest: combining the predictions of multiple independent decision tree models. J. Chem. Inf. Comput. Sci 2003. [12] Feng Q, Dueva E, Cherkasov A, Ester M. Padme: A deep learning-based framework for drug-target interaction prediction. arXiv preprint arXiv:1807.09741 2018. [13] https://www.analyticsvidhya.com/blog/2017/09/common-machine-learning-algorithms/ [14] L. Burggraaff, P. Oranje, R. Gouka, P. van der Pijl, M. Geldof, H.W. van Vlijmen, A.P. IJzerman, G.J. van Westen, Identification of novel small molecule inhibitors for solute carrier sglt1 using proteochemometric modeling, Journal of cheminformatics 11 (1) (2019). [15] D.S. Wishart, Y.D. Feunang, A.C. Guo, E.J. Lo, A. Marcu, J.R. Grant, T. Sajed, D.Johnson, C. Li, Z. Sayeeda, et al., Drugbank 5.0: a major update to the drugbank database for 2018, Nucleic acids research 46 (D1) (2018). [16] A. Alimadadi, S. Aryal, I. Manandhar, P.B. Munroe, B. Joe, X. Cheng, Artificial intelligence and machine learning to fight covid-19 (2020). [17] J.B. Cross, Methods for virtual screening of gpcr targets: Approaches and challenges, in: Computational Methods for GPCR Drug Discovery, Springer,2018. [18] K. Zhao, H.-C. So, Using drug expression profiles and machine learning approach for drug repurposing, in: Computational methods for drug repurposing, Springer, 2019. [19] Nicolas, J., Artificial intelligence and bioinformatics, in A Guided Tour of Artificial Intelligence Research. 2020. [20] Kohli, A., et al., Concepts in US Food and Drug Administration regulation of artificial intelligence for medical imaging. American Journal of Roentgenology, 2019. [21] Zhou, Y., et al., Artificial intelligence in COVID-19 drug repurposing. The Lancet Digital Health, 2020. [22] Jiménez-Luna, J., F. Grisoni, and G. Schneider, Drug discovery with explainable artificial intelligence. Nature Machine Intelligence, 2020. [23] Mohanty, S., et al., Application of Artificial Intelligence in COVID-19 drug repurposing. Diabetes & Metabolic Syndrome: Clinical Research & Reviews, 2020. [24] Paul, D., et al., Artificial intelligence in drug discovery and development. Drug Discovery Today, 2020. [25] Cano G, Garcia-Rodriguez J, Garcia-Garcia A, Perez-Sanchez H, Benediktsson JA, Thapa A, Barr A Automatic selection of molecular descriptors using random forest: Application to drug discovery (2017). [26] Chen H, Zhang Y, Kalra MK, Lin F, Chen Y, Liao P, Zhou J, Wang G Low-dose ct with a residual encoder-decoder convolutional neural network (2017). [27] Mei J-P, Kwoh C-K, Yang P, et al. Drug–target interaction pre-diction by learning from local information and neighbors. Bioinformatics 2012. [28] You J, McLeod RD, Hu P. Predicting drug–target interac-tion network using deep learning model. Comput Biol Chem 2019.
Copyright
Copyright © 2023 Niharika Gupta, Priya Khobragade. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.

Download Paper
Paper Id : IJRASET52112
Publish Date : 2023-05-12
ISSN : 2321-9653
Publisher Name : IJRASET
DOI Link : Click Here
 Submit Paper Online
Submit Paper Online