

Ijraset Journal For Research in Applied Science and Engineering Technology
NGS and Mutational Profile Analysis of Non-Small-Cell Lung Carcinoma (NSCLC)
Authors: Uma Kumari, Nidhi Agrawal
DOI Link: https://doi.org/10.22214/ijraset.2023.50880
Certificate: View Certificate
Abstract
Computational analysis imitates the functioning of biological information systems to anticipate and offer novel perspectives on the biological realm. Genome sequencing analysis interactive techniques with the aim of improving the standards of cancer patient, has progressed greatly over the last decades, and is now considered to be one of the major scientific fields that promote innovation in the technology and healthcare sectors. In the field of bioinformatics, the first widespread application of data analysis was the creation of the first collection of systematic data representing the genome sequences of the eukaryotic organisms, termed as the eukaryotic genome, or the \"de novo\" genome. Tumor suppressor proteins are a class of proteins that are involved in preventing cancer from occurring. Several tumor suppressor proteins have been found to be mutated in certain forms of cancer, resulting in the loss of their tumor suppressing properties. Lung cancer develops when normal lung cells change, or mutate, in a way that alters their natural growth and death cycle, resulting in unregulated cell division that produces too many cells.Deep sequence analysis of non-small cell lung cancer and integrated analysis of gene expression. Understanding the biology of cancer is required to improve patient outcomes. Next-generation sequencing (NGS) is a powerful tool for whole genome characterization, enabling comprehensive exploration and understanding of the complex interactions between genes, proteins and their environment. NGS in the field of oncology is still in its infancy and the wealth of data that it is capable of generating is revolutionizing our understanding of cancer. ORFs have led to many exciting discoveries in the field of oncology, including the identification of tumor suppressor proteins. One of the most exciting recent discoveries is in the field of tumor suppressor proteins. Tumor suppressor proteins are proteins that play a role in the regulation of cell proliferation and division. Mutations in certain tumor suppressor genes cause cancer, and as a result, a great deal of research has gone into the identification of new tumor suppressor genes.
Introduction
I. INTRODUCTION
Medical informatics is a basic biomedical science that has a wide variety of application areas which involve improvements in the management of any information relevant to patient care and community health. Onco-informatics is an application science under this broad field. By the help of data analysis has enabled the collection of a huge amount of information in the area of oncology. Several oncology-specific databases and blogs that are clinically relevant are described in Oncology paper. Bioinformatics database and software to identify genes, establish their functions, and develop gene-based strategies for preventing, diagnosing, and treating disease. Genome sequencing reaction produces DNA sequence that is several hundred bases long. Combination tools and technique work in use how Gene sequences technique typically run for thousands of bases. To increase survivals of patients, it is important to explore new biomarkers for diagnosing, subtyping, and prognosing of NSCLCs. More studies have been focused on ncRNAs, particularly miRNAs in the past few years [1]. Many lncRNAs are reported as candidate biomarkers, e.g., highly up-regulated in liver cancer (HULC) in human hepatocellular carcinoma and prostate cancer gene 3 (PCA3) in prostate cancer. MALAT1[2] XIST, and HIF1A-AS1[3] are overexpressed in NSCLC patients’ serum when compared with controls. These lncRNAs may act as diagnosis biomarkers for screening NSCLCs via peripheral blood detection. White et al was able to identify 27 lung cancer-associated lncRNAs, which can be used as novel biomarkers for stratifying LADs and LSCCs [4]. Currently, surgical removal of tumor, chemotherapy, and chest radiotherapy and target therapy are used alone or in combination to treat patients with NSCLC [5]. Due to drug resistance many drug therapies fail. Studies showed that there is an association between the expression of certain lncRNAs and chemotherapeutic sensitivity of cancer cells. For example, H19 induced P-glycoprotein- and MDR1-associated drug resistance in liver cancer cells [6]. Resistance to cisplatin, carboplatin, and EGFR-TKIs is inevitable in treating NSCLCs [7]. In lung cancer deregulation of TC21 can trigger cellular transformation and contribute to human oncogenesis because similar signal transduction pathway is used by oncogenic Ras[8]. Recent studies show that Rabl3 is a novel oncogene which regulates the oncological behaviour of human cancer cells.
Rabl3 may be a controlling agent in tumorigenesis and a new target for anti-tumor treatment [9]. Translational profile analysis Chromosome 9 open reading frame 86 (C9orf86), also known as Rab-like protein 1 (RBEL1), is located at 9q34.3. C9orf86 was overexpressed in NSCLC cell lines.C9orf86, also known as RBEL1, is a novel Ras superfamily protein. For data analysis work ras family proteins play an important role in tumorigenesis, by promoting tumor growth and invasion and regulating processes such as cell cycle progression, proliferation, apoptosis, migration, and survival. High rates of KRAS-activating missense mutations have been detected in many cancers, including NSCLC. Environmental and occupational exposures well as genetic susceptibility [10] are major to contributor to the lung cancer risk in non-smokers. Inhibitors of epidermal growth factor receptor (EGFR) tyrosine kinase (TK), gefitinib and erlotinib, have shown substantial activity in patients whose tumor cells harbor specific mutations in the EGFR TK domain. Structural analysis EGFR TK domain mutations and fusion kinases involving EML4-ALK are present more often in the tumor specimens from life-long never smokers than from smokers [11]. Coding exons sequencing of 623 candidate cancer genes in 188 lung adenocarcinomas identified 26 significantly mutated genes in lung adenocarcinoma, consisting of a set of oncogenes (EGFR, KRAS, ephrin receptor genes, ERBB4, KDR, FGFR4 and NTRK genes) [12] and tumor suppressors (TP53, STK11, NF1, RB1, ATM and APC)[13]. Particularly, C:G→A:T transversions were present predominantly in tobacco smokers whereas C:G→T:A transitions were the most frequent type of point mutations in non-smokers with lung cancer and the former light smoker[14]. PyMOL is written in Python, which is one of the most popular programming languages, it can be extended to Python plugins as well [15]. PyMOL can be used for visualization and enhanced analysis functions. The computational drug discovery function of PyMOL has been successfully applied to ?nd new drug candidates for various targets. Macromolecular Visualization of molecules is the preliminary task ofCADD [16]. PyMOL has been widely used for 3D visualisation of macromolecules and it becomes one of the most popular tools for preparing high-resolution images of macromolecules for publications [17].
II. MATERIAL AND METHODS
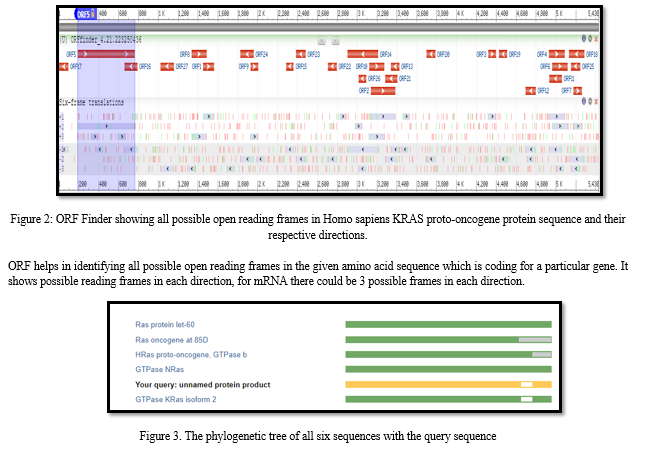
The basic tool in bioinformatics is a computer program that mimics the way biological information systems function in order to predict and provide new insights into the biological world. The program performs computations that provide biologists with information about DNA sequences, protein structures, gene regulatory mechanisms, and other biological structures and processes. The use of computer software in bioinformatics is the basic underlying principle of the field. The study utilized the National Center for Biotechnology Information (NCBI) to obtain the sequences. Biological databases related to biotechnology and biomedicine, and provides a variety of information when using bioinformatics tools and services. Examples of such resources include GenBank, which contains DNA sequences, and PubMed. Development of computational tools has made it possible to identify new opportunities in the field of bioinformatics. Currently, the market is driven by the increasing demand for tools for drug discovery, population genetics, and proteomics. The complex and heterogeneous nature of the biological data collected and generated in modern biological laboratories has led to the proliferation of tools and applications to identify, capture, describe, analyse, and manipulate biological data. These tools are designed to provide a rational platform to resolve the multitude of challenges that are faced in modern biological research. Following this, ORF Finder was used to scans the sequence for open reading frames (ORFs). Through this ORF range as well as the protein translation for each ORF was found.
III. RESULTS AND DISCUSSION
After obtaining the protein sequence for the KRAS proto-oncogene, from NCBI, it is transformed into the FASTA format and utilized as a search sequence in BLAST. BLAST is able to analyze a protein sequence by comparing it to a database that contains other protein sequences. The outcome of the BLASTP search displayed a list with hundreds of sequences in the description that were comparable to the sequence that was being queried for.
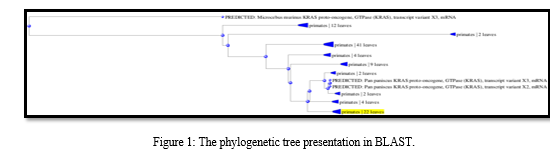
Pylogenetic Sequence tree presentation an implicit between the database sequences is construct based upon the alignment of those (database) sequence to the query. The sequence was scanned for open reading frames (ORFs) using ORF Finder, which enabled the determination of the range for each ORF as well as the protein translation for each ORF.
From left to right, the output displays the phylogenetic tree of all six sequences with the query sequence. The query sequence is colored yellow, and the matching sequences are green (from the reference database). In the multiple sequence alignment, white gaps represent deletions, while regions that did not align with the query sequences are marked in grey. The constraint-based multiple sequence alignment method was used, which involved retrieving the query sequence from NCBI in FASTA format and performing multiple alignments through COBALT. COBALT doesn't aim to employ all constraints but instead utilizes a high-scoring subgroup that can vary as the alignment progresses. The result obtained from COBALT highlights the amino acid residues found in the sequences, providing us with an understanding of which amino acid is present in the common sequences.
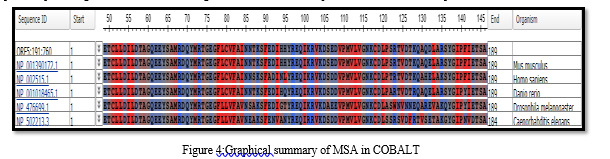
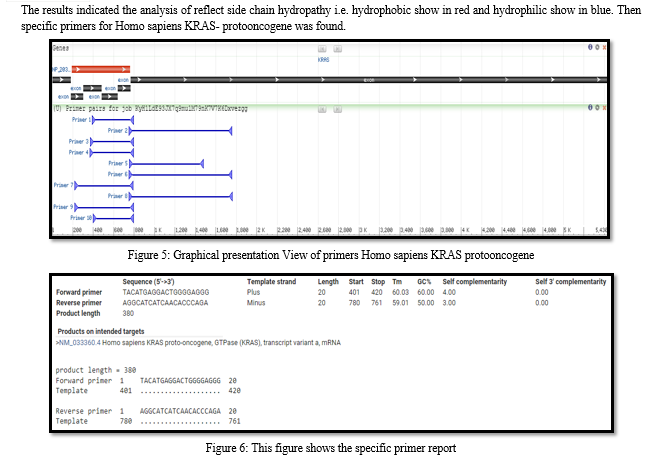
Conclusion
In mutational profile analysis of alignment obtained and stable region and unstable which indicates the position where the mutation occurs, the system network topology that produced the phylogenetic tree of lung cancer epidemic distance method, and system area networks mutation. Lung cancer lines have to pharmacological and other biomedical companies, demonstrating their usefulness for therapeutic and other translational applications. ORF to mean any contiguous stretch of codons beginning with a start codon, ending with a stop codon, and with no intermediate in-frame stop codons, ORF to be a “protein-coding gene” if it is translated into a functional protein, by which we mean a protein that contributes to viral/ transmission, replication. Bioinformatics tools employed for identifying sequence highly similar to those in the small cell lung cancer.
References
[1] Zeringer E., Rai A.J., Decastro J. et al, “Abstract 3387: A complete workflow for high throughput isolation of serum microRNAs and downstream analysis by qRT-PCR: application to cancer biomarker discovery”, Cancer Research, 75, 3387-3387, 2015 [2] Weber DG, Johnen G, Casjens S, Bryk O, Pesch B, Jöckel KH, Kollmeier J, Brüning T, “Evaluation of long noncoding RNA MALAT1 as a candidate blood-based biomarker for the diagnosis of non-small cell lung cancer”, BMC Res Notes, 6;6:518, Dec, 2013. [3] Tantai J, Hu D, Yang Y, Geng J. “Combined identification of long non-coding RNA XIST and HIF1A-AS1 in serum as an effective screening for non-small cell lung cancer”, Int J ClinExpPathol, 8(7):7887-95, Jul 2015 [4] White NM, Cabanski CR, Silva-Fisher JM, Dang HX, Govindan R, Maher CA, “Transcriptome sequencing reveals altered long intergenic non-coding RNAs in lung cancer”, Genome Biol., 13;15(8):429, Aug, 2014 [5] Albain KS, Swann RS, Rusch VW, Turrisi AT 3rd, Shepherd FA, Smith C, Chen Y, Livingston RB, Feins RH, Gandara DR, Fry WA, Darling G, Johnson DH, Green MR, Miller RC, Ley J, Sause WT, Cox JD, “Radiotherapy plus chemotherapy with or without surgical resection for stage III non-small-cell lung cancer: a phase III randomised controlled trial”, Lancet, 1;374(9687):379-86, Aug, 2009. [6] Tsang WP, Kwok TT, “Riboregulator H19 induction of MDR1-associated drug resistance in human hepatocellular carcinoma cells”, Oncogene, 19; 26(33):4877-81, Jul, 2007. [7] Schneider-Merck T, Pohnke Y, Kempf R, Christian M, Brosens JJ, Gellersen B, “Physical interaction and mutual transrepression between CCAAT/enhancer-binding protein beta and the p53 tumor suppressor”, J BiolChem, 281(1):269-78, Jan, 2006. [8] Graham SM, Cox AD, Drivas G, Rush MG, D\'Eustachio P, Der CJ, “Aberrant function of the Ras-related protein TC21/R-Ras2 triggers malignant transformation”, Mol Cell Biol., 14(6):4108-15, Jun 1994. [9] Li Q, Wang L, Zeng L, Zhang Y, Li K, Jin P, Su B, Wang L, “Evaluation of the novel gene Rabl3 in the regulation of proliferation and motility in human cancer cells”, Oncol Rep., 24(2):433-40, Aug, 2010. [10] Sellers TA, Bailey-Wilson JE, Elston RC, Wilson AF, Elston GZ, Ooi WL, Rothschild H, “Evidence for mendelian inheritance in the pathogenesis of lung cancer”, J Natl Cancer Inst, 1;82(15):1272-9, Aug, 1990. [11] Soda M, Choi YL, Enomoto M, et al., “Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer”, Nature, 448(7153):561-566, 2007. [12] Virk N and Kumari U, “Genome Sequence Analysis of Lungs Cancer Protein WDR74 (WD Repeat-Containing Protein)”, IJRASET, 10(V):4533-4537, 2022 [13] Ding L, Getz G, Wheeler DA, et al., “Somatic mutations affect key pathways in lung adenocarcinoma”, Nature, 455(7216):1069-1075, 2008. [14] Lee W, Jiang Z, Liu J, et al., “The mutation spectrum revealed by paired genome sequences from a lung cancer patient”, Nature., 465(7297):473-477, 2010. [15] Kumari Uma and Choudhary, Ashok Kumar, “Genome Sequence Analysis of SolanumLycopersicum by Applying Sequence Alignment Method to Determine the Statistical Significance of an Alignment”, International Journal of Bio-Technology and Research (IJBTR), 2249-6858;9-12, 2016. [16] Kumari Uma, &Choudhary, A. K, “Genome sequence analysis of solanumlycopersicum showing the phylogenetic relationship based on multiple sequence alignment and conserved domain proteins”, International journal of advanced biotechnology and research, 7(4), 2012-2014, 2016. [17] Vinita Kukreja, Uma Kumari, \"Genome Annotation of Brain Cancer and Structure Analysis by applying Drug Designing Technique\", International Journal of Emerging Technologies and Innovative Research, 9(5);473-k479, May, 2022.
Copyright
Copyright © 2023 Uma Kumari, Nidhi Agrawal. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.

Download Paper
Paper Id : IJRASET50880
Publish Date : 2023-04-23
ISSN : 2321-9653
Publisher Name : IJRASET
DOI Link : Click Here
 Submit Paper Online
Submit Paper Online