

Ijraset Journal For Research in Applied Science and Engineering Technology
Predicting Cancerous Genes through an In-Silico Approach
Authors: Ipsita Saha, Kriti Ghosh, Srabani Kundu, Amrut Ranjan Jena, Moloy Dhar
DOI Link: https://doi.org/10.22214/ijraset.2022.44999
Certificate: View Certificate
Abstract
In Sequence alignment, the chains of DNA, RNA, or protein are aligned and compared to recognize homogeneous portions which could be the result of functional, organizational, or evolutionary connections between the chains. This process gives similar sequences as its result. Similar sequences often have the same biological function. This knowledge can be used to draw further conclusions that we can ascertain the function of a query DNA sequence by comparing it with a target sequence performing a known function. In this article, we use this idea to develop an Advanced Java-based web application where the user interface allows the user to enter an unknown DNA sequence. Using pattern-matching algorithms, the application can calculate how similar the sequence is with genes stored in a database. Based on the result, a trained medical practitioner will be able to determine whether the unknown gene can be cancerous.
Introduction
I. INTRODUCTION
Cells, the constituent elements of life, contain a plethora of information stored in the long nucleotide chains called DNA. This information, when analyzed properly can shed light on the reason behind different life-threatening diseases. As the study of Genetic information is crucial to understanding complicated diseases like Cancer, it is evident that the exploration of DNA, RNA, or protein sequences is extremely important. However, as the genetic information is stored in the thousands of nucleotide-long DNA chains, manual examination and interpretation of the genetic materials are not convenient. Computational tools and techniques on the other hand can do the job more efficiently [1].
Sequence Alignment is one such computational technique where DNA, RNA, or protein sequences are aligned to point out conserved regions that may have a functional, structural, or evolutionary correlation. Sequence Alignment results give similar sequences, i.e., sequences having similar functions. Hence, we can establish the function of a query DNA sequence by comparing it with a target sequence performing a known function. This approach is extremely useful when studying different life-threatening diseases like Cancer. In fact, in the diagnosis and treatment of cancer, this can play a huge role. Our approach is to prepare a web application where the user will be able to enter an unknown DNA sequence. Using pattern-matching algorithms like Needleman-Wunsch [3], Smith-Waterman [2], etc., the application will calculate the sequence similarity with genes stored in a local database. Based on the result, a trained practitioner will be able to say whether the unknown gene can be cancerous.
The flow of this research article is as follows:
- At first, we have discussed a few widely used sequence alignment algorithms and their shortcomings in section 2.
- We have then introduced our chosen algorithm and the advantages it possesses. Also, we have discussed the modifications that we have made.
- Then we have described the architecture we have followed in our proposed web application.
- We have discussed our results and future scope in the conclusion.
II. BACKGROUND AND RELATED WORK
The genetic materials in cancerous cells go through a series of changes that modify the normal cell properties [8]. Over time,genes become mutated more frequently, and cells develop different properties like malignancy, anaplasia, metastasis, loss of adhesion [9], etc. Mutated genes are often linked with different forms of cancer. Genes can be classified into three groups: the proto-oncogenes, which normally enhance cell division, and the tumor suppressor genes that prevent cell division. The third group is DNA repair genes which help avert mutations that lead to cancer. In a healthy state, these genes maintain the balance. Once mutated they disrupt the system and may produce cancer. If homology or shared ancestry is found between a known cancerous gene and an unknown query sequence, the query sequence may be transformed into a cancerous gene through mutations. Sequence alignment can help in finding homology.
There are different classes of sequence alignment algorithms namely global alignment algorithms and local alignment algorithms. Global alignment algorithms align each residue in all the sequences. They are suitable for sequences similar in length. Meanwhile, local alignment algorithms find regions where the density of similarity is high. These techniques use different scoring mechanisms to score matches, where the residue at a particular index is identical in both the sequences under observation and gaps, where a residue seems to be deleted in one sequence and inserted in the other. The resultant score is then used to ascertain the similarity among the sequences. A few popular algorithms are:
A. Needleman-Wunsch Algorithm
It is a global alignment algorithm. Needleman Wunsch algorithm [3] finds the ideal alignment between two similar protein or nucleotide sequences of roughly equal length. This global alignment algorithm is a dynamic programming approach to finding the optimal alignment of two sequences in their entirety among many possible alignments. This algorithm uses a scoring matrix which helps in verifying the similarity between the sequences and a traceback method of the matrix provides the optimal alignment. However, in the case of highly dissimilar sequences, the alignment may not have any biological significance.
B. BLAST
BLAST is a local alignment algorithm that is one of the most extensively used alignment algorithms in the world. It is a complex tool that conducts similarity searches against different sequence databases to provide related sequences. It is relatively faster than its competitors. However, it is a heuristic algorithm. As a result, it does not consider all the alignments that it finds.
III. PROPOSED MODELING
We have developed a web application based on advanced java that provides the similarity between a query gene and the cancerous genes stored in a local database. This portion of the article is organized in two sub-parts:
A. Software Architecture
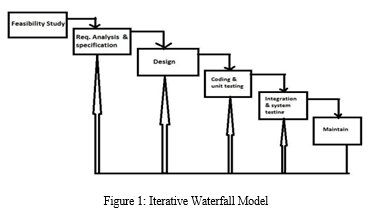
We have developed our web application following the iterative waterfall model [11,12] and MVC architecture. In the Iterative waterfall model [Figure1], each phase of software development is linearly conducted one after another. Moreover, there is a feedback path from every stage of the lifecycle to its previous phase so that one can redirect their course of action.
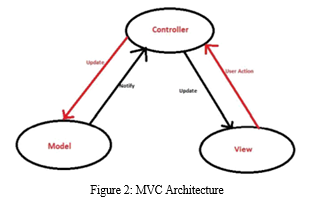
The architecture of the software is based on MVC or Model-View-Controller architecture [13,14]. It is a three-tier architecture that comprises a model object, a controller, and a view.
View provides the visualization of the data included in the model to the user. In this case, the user interface of our application is easy to operate. It allows the user to enter the query sequence with ease.
A model is an object which contains the data. In this case, it is the local database containing cancerous gene sequences. The model also contains the logic to revise the controller if the data is modified.
The controller controls the flow of data into the model and modifies the view accordingly.
The advantage of this architecture is that it keeps the model part and the view part separate which makes data manipulation easier. The model is explained in Figure 2.
B. Proposed Algorithmic Logic
We have developed a web application called SeQALign-C which is grounded in the local alignment algorithm Smith-Waterman algorithm. This application has an easy-to-use user interface through which the user can enter the query DNA sequence. After the data has been enteredthis query sequence is utilized to perform alignment with the cancer gene sequences stored in the local database. After the database is exhausted, the alignment similarity scores are calculated as mentioned in the flowchart in Figure 3.
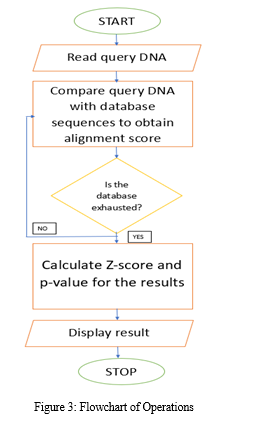
The Smith-Waterman algorithm [2] is a dynamic programming approach that divides problems into subproblems to combine the solutions to these smaller problems to develop the solution of the original problem [5]. The algorithm has three phases:
- Initializing the score matrix
- Scoring
- Traceback

where A is the gap penalty [7]. The Traceback procedure is initiated from the maximumvalue and then the path followed to reach there is traced back in the reverse direction and the alignment is obtained.
Now the highest value in the score matrix is the similarity or match score. However, a match score alone is not a good measure of how significant a particular match is. It is essential to establish whether an alignment has any biological significance [10]. As a result, we propose the application of the Smith-Waterman algorithm along with a few statistical measures like z-score and p-value. Z-score represents how many standard deviations above or below the mean of the population a particular alignment lies. The proposed approach is portrayed in the following algorithm.

IV. RESULTS AND DISCUSSIONS
The operation of the web application is very straightforward. The local alignment algorithm chosen for the application is the Smith-Waterman algorithm. It is a local alignment algorithm. Although it is not as fast as its heuristic counterpart BLAST, it is less likely to miss important regions of similarity [6]. As our objective is the prediction of cancerous patterns in gene sequences, we are more concerned with precision than time consumption. Moreover, the inclusion of statistical measures ensures that a medical practitioner will be able to resolve whether the query sequence is similar to any sequence in the local database and whether the similarity is significant.
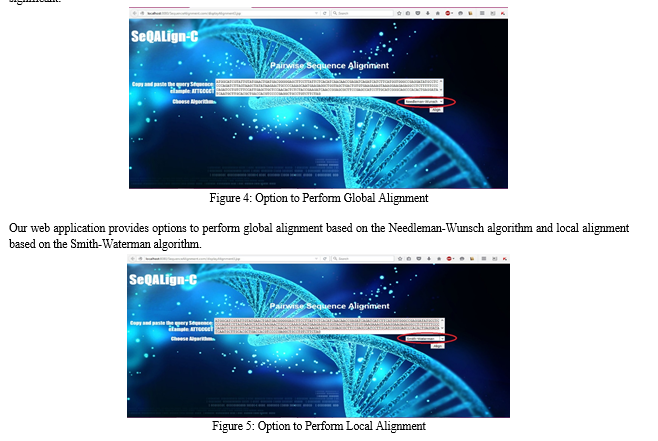
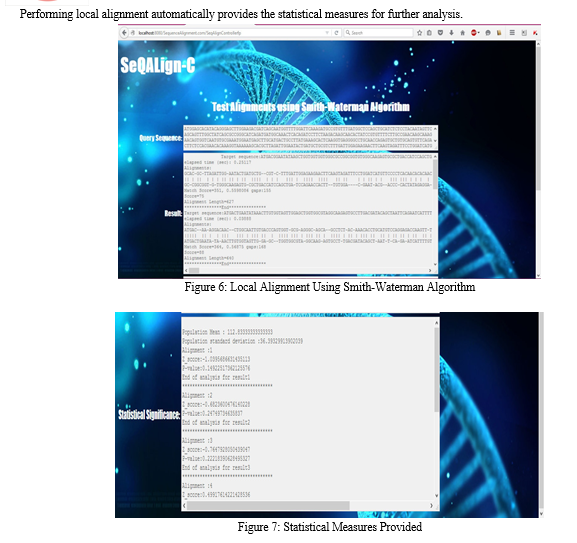
V. FUTURE SCOPE
In the future, the application can be made more comprehensive by making it compatible with online cancer databases. Moreover, it can also be extended towards the prediction of other diseases like Thalassemia. This software could also be incorporated into a bigger project, where a novel gene could be used as a query sequence, and after obtaining the result, gene expression data could be used to analyze the effect of the gene. In the future enhanced version of the software, a 3D protein viewing model can be added to determine the structure of the protein coded by the novel gene.
Conclusion
Homology is the existence of shared ancestors between gene sequences and often homology can be found in similar sequences. Thus, sequence similarity can be used to determine the function or structure of a query gene. If a query gene is sufficiently similar to a known cancerous gene sequence, then it will mean that through mutations the query gene can be transformed into an oncogene which in turn can cause cancer. To determine the significance of the similarity statistical measures like z-score and p-values are provided. In the future, this application can be further developed to help in the diagnosis and treatment of cancer.
References
[1] Waterman, Michael S. Introduction to computational biology: maps, sequences and genomes. Chapman and Hall/CRC, 2018. [2] Smith, Temple F., and Michael S. Waterman. \"Identification of common molecular subsequences.\" Journal of molecular biology 147, no. 1 (1981): 195-197. [3] Needleman, Saul B. &Wunsch, Christian D. (1970). \"A general method applicable to the search for similarities in the amino acid sequence of two proteins\". Journal of Molecular Biology. 48 (3): 443–53. doi:10.1016/0022- 2836(70)90057-4. PMID 5420325. [4] Pearson, William R. \"An introduction to sequence similarity (“homology”) searching.\" Current protocols in bioinformatics 42, no. 1 (2013): 3-1. [5] Cormen, T. H.; Leiserson, C. E.; Rivest, R. L.; Stein, C. (2001), Introduction to Algorithms (2nd ed.), MIT Press & McGraw–Hill, ISBN 0-262-03293-7 . pp. 344. [6] \"Bioinformatics Explained: BLAST versus Smith-Waterman\" (PDF). 4 July 2007. [7] Wikipedia,Gap penalty, 2016. url: https://en.wikipedia.org/wiki/Gap_penalty. [8] National Cancer Institute. What is Cancer? url: http : / / www . cancer.gov/about-cancer/what-is-cancer. [9] Bhakta Chaudhury Bardhan. A Text Book of Biology. 2009, pp. 81- 83. [10] Mitrophanov, Alexander Yu, and Mark Borodovsky. \"Statistical significance in biological sequence analysis.\" Briefings in Bioinformatics 7, no. 1 (2006): 2-24. [11] Petersen, Kai; Wohlin, Claes; Baca, Dejan (2009). Bomarius, Frank; Oivo, Markku; Jaring, Päivi; Abrahamsson, Pekka (eds.). \"The Waterfall Model in Large-Scale Development\". Product-Focused Software Process Improvement. Lecture Notes in Business Information Processing. Berlin, Heidelberg: Springer: 386–400. doi:10.1007/978-3-642-02152-7_29. ISBN 978-3-642-02152-7. [12] Reenskaug, Trygve; Coplien, James O. (20 March 2009). \"The DCI Architecture: A New Vision of Object-Oriented Programming\". Artima Developer. Archived from the original (html) on 23 March 2009. Retrieved 3 August 2019. [13] Davis, Ian. \"What Are The Benefits of MVC?\". Internet Alchemy. Retrieved 2016-11-29.
Copyright
Copyright © 2022 Ipsita Saha, Kriti Ghosh, Srabani Kundu, Amrut Ranjan Jena, Moloy Dhar. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.

Download Paper
Paper Id : IJRASET44999
Publish Date : 2022-06-28
ISSN : 2321-9653
Publisher Name : IJRASET
DOI Link : Click Here
 Submit Paper Online
Submit Paper Online