

Ijraset Journal For Research in Applied Science and Engineering Technology
Review on Processes in Liquid-Liquid and Solid Phase Extraction
Authors: Dr. Aghogho Blessing Obukohwo
DOI Link: https://doi.org/10.22214/ijraset.2023.48272
Certificate: View Certificate
Abstract
The process of separating a component of mixture of liquid using liquid solvent is known as solvent extraction. The component being separated is completely insoluble in the solvent known as the carrier liquid. Distribution coefficient and partition coefficient are used to quantitatively determine the degree of solubility of a solute in a solvent compared to its solubility in another solvent. In liquid-liquid extraction (LLE) the solvents used should have maximum transfer of solute from carrier into the solvent. The solvent used must have high affinity for the solute to be extracted and it must not be completely miscible with the carrier liquid. Solid phase extraction (SPE) is a solvent-extraction system with a stationary phase by adsorption onto a solid support (usually silica) and the other liquid phase which is mobile. It uses small membranes or columns and many of the extracting agents used in LLE are also used in SPE. SPE generates less amount of wastes and it is an excellent substitute for LLE as it is faster and more efficient. It requires samples as small as 50 – 500ul and relatively small volumes of solvents not as pure as may be required by LLE. The working principles of SPE are like that of LLE were partitioning takes place between two immiscible liquids but in SPE the analytes to be extracted are partitioned between liquid and solid. This paper has reviewed the various processes involved in solvent extraction considering diluents in solvent extraction, liquid-liquid extraction and solid-phase extraction.
Introduction
I. INTRODUCTION
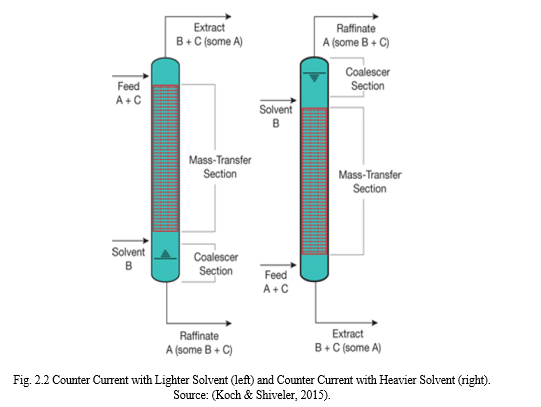
Solvent extraction involves the use of a liquid solvent to remove or separate a liquid component from a mixture of liquid. It is a selective removal of a solute using solvent from a liquid mixture. The component in transition is completely taken by the solvent and it is assumed that there is an absolute insolubility in the carrier liquid. Separation process occurs in two phases; the first is the extract phase where A and B mixes and C is the residue while at the Raffinate phase A and B becomes the residue and C is the main component of interest. To obtain a pure component of interest a process known as rectification is carried out. In this process, the component in transition is separated from the solvent. Fig 2.2 shows the principles of liquid – liquid extraction. The solvent can be recirculated for extraction again (Chizhkov et al., 2011; Yang et al, 2013; Mulle et al., 2015; Hamburg, 2017).
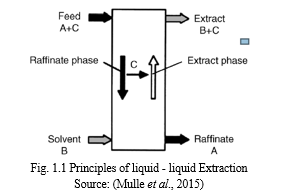
Solvent extraction is carried out in an extraction column or a mixer settler equipment which has a rotating internal part used to create droplet sizes. The droplets formed in the dispersed phase then accumulate below or above the continuous phase depending on the different relative densities of both liquids. A boundary is formed between the dispersed and the continuous phase which is referred to as an interface.
This interface may be located at the bottom or top of the extraction column. A typical extraction unit has two inlet streams made up of the liquid carrier and the solvent; it contains the outlet stream made up of the raffinate and solute-rich extract. Solvent extraction has a well-established theory and it is a continuous separation method. The following are metals recovered by solvent extraction techniques: cobalt, nickel, thorium, uranium, platinum group metals, tungsten, molybdenum, lanthanides, copper, hafnium, zirconium, tantalum, beryllium, boron, niobium (Hamburg, 2017; Tarihi, 2001; Marlap, 2004; Demim et al, 2013a; Demim et al, 2013b; Yang et al, 2013; Demim et al, 2014; Mulle et al., 2015; Koch & Shiveler, 2015; Leyma et al, 2016).
In the dispersed phase mass transfer would occur between droplets and surrounding liquids in the continuous phase. However, one property both solvents must possess for separation to occur is difference in density; the solute can either be a solid or liquid while the extracting solvent can be water, water-miscible or water-immiscible. (Marlap, 2004; Yang et al, 2013; Koch & Shiveler, 2015).
II. DISTRIBUTION COEFFICIENT
Distribution coefficient and partition coefficient, Kd is described as the different solubility of a solute in the solvent pairs of an extraction system and it is used to describe solvent extraction quantitatively. The coefficient represents the solute solubility in one solvent relative to its solubility in another solvent and it is known as the equilibrium constant. The concentration of the equilibrium a one phase also has a direct relationship with the concentration of the solute in the other phase after equilibrium has been reached. Distribution Coefficient Kd can be expressed as:
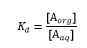
Where [Aorg] is the concentration of solute in organic phase and [Aaq] is the concentration of solute in aqueous phase while Kd is the distribution coefficient constant. The unit of the concentration is usually in molality (g/g or moles/kg) units and this makes the constant unitless. Saturated concentrations for the solute in each solvent is represented by the solubility which varies with temperature; this makes the coefficient temperature-dependent but not by a factor which is constant. For instance, when a solute in hexane / water system has a distribution coefficient of 90 it means that at saturation conditions the solute is 90 times more soluble in hexane solution than it is in water. However, the water may also contain small amount of the solute. The solute is selectively dissolved in one solvent, but it is not completely removed from the other solvent. In the example of hexane / water system above if the coefficient is large then it indicates that more solutes would be distributed in hexane after extraction while a small quantity would remain in the water. Repeated extractions are used in solvent extraction procedures to extract a solute quantitatively from a liquid mixture. The distribution law expression is not thermodynamically rigorous, and it does not account for instances where the solute is involved in a chemical reaction like association or dissociation in either phase. However, the distribution law expression is a useful approximation (GBH, no date; Marlap, 2004; Yang et al, 2013; Xie et al, 2014).
A. Separation Factors
The separation or selectivity factor, S, can be represented as:
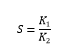
Where 1= component to be extracted
2 = component to be retained
The separation of K1 from K2 increases as the selectivity factor, S, increases. When the value of S is high it shows that there is a high degree of extraction with a few number of extraction stages approximation (GBH, no date; Marlap, 2004; Yang et al, 2013 Xie et al, 2014).
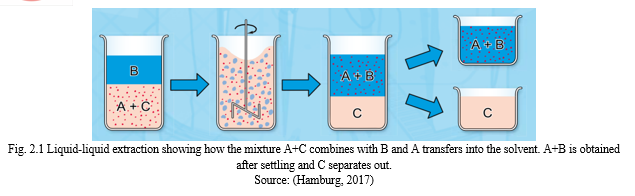
Solvent Extraction is dependent on solute distribution between solvent and feed streams. The separation or selectivity factor and distribution coefficient are two important parameters the number of extraction stages. It is important for solvents used in solvent extraction to have high distribution coefficient as this will help to reduce the rate of solvent circulation. Fig. 2.3 shows how the mixture A+C combines with B and A transfers into the solvent (Tarihi, 2001). High distribution coefficient or high value of k in solvents used for extraction would also reduce the degree of solute removed during regeneration of the solvent. Also, water contamination would be low as this will reduce loss of solvent. However, the k value is not dependent on the concentration of the solute and the Extract to Raffinate phase ratio (GBH, no date; Tarihi, 2001; Baba et al., 2011).
B. Mode of Operation of Extraction
Extraction is operated in either a co-current mixing mode or a counter current mixing mode. This process is in staged units which is continuous. In the co-current mixing mode, there is one theoretical stage per extraction unit while the counter-current mixing mode is open to multiple stages per extraction unit. However, as the counter-current mixing is open to multiple stages of extraction it is usually preferred over the co-current mixing. The counter-current extractor could be arranged in two different ways depending on the density of the solvent in relation to the solute carrier. In the case of a solvent less dense than the carrier the solvent is fed through the bottom of the column as seen in Fig 2.4 (left) and as the solvent moves upward the solute is carried along to the top of the extractor while the carried liquid is removed from the. For a solvent denser than the carrier the solvent is fed through the top of the column Fig 2.4 (right) and as the solvent moves downward the solute is carried along to the bottom of the extractor (Baba et al., 2011; Chizhkov et al., 2011; Koch & Shiveler, 2015).
- Characteristics of Solvents for Liquid – Liquid Extraction
In liquid-liquid extraction the solvents used should have maximum transfer of solute from carrier into the solvent. The solvent used must have high affinity for the solute to be extracted and it must not be completely miscible with the carrier liquid. Ideal solvent for liquid-liquid extraction should:
a. Be nonreactive with other chemicals involved in the extraction process.
b. Have a density difference of 150 kg/m3 between the carrier liquid.
c. Have high boiling point for handling material easily.
d. Have low solubility for the carrier liquid and high solubility for the solute.
e. Have a mid-level interfacial tension of 5 – 30 dyne /cm.
f. Have high resistance to thermal degradation.
g. Low viscosity for easy handling.
h. Be relatively inexpensive.
i. Be non-volatile, non-flammable, nontoxic or corrosive to the process equipment.
j. Distribution or Partition Coefficient.
k. Stable throughout three principal stages
l. Have high extraction, scrubbing and stripping rates.
m. Have high loading capacity.
(GBH, no date; Chizhkov et al., 2011; Koch & Shiveler, 2015)
A comparison study for the most suitable solvent may be required as it is difficult to find a solvent with all the criteria as listed above. However, it might be suitable to design a solvent of interest to possess the desired combination of selectivity and partition coefficient. For example, the pH in a chemical solvent may be adjusted to achieve influence the activity of the solvent. Also, a solvent might be designed or modified to remove a solute from a feed and then release it into another solvent when the pH is altered either by the addition of an acid or alkali. The extract phase or raffinate could be made continuous depending on the stability of the interface in the mixture which inhibits settling but improves mass transfer (GBH, no date; Baba et al., 2011; Chizhkov et al., 2011; Koch & Shiveler, 2015).
2. Application of Solvent Extraction
Liquid – liquid extraction is employed in industrial separation processes. However, it is likely that solvent extraction would be very useful separation method in future as some industrial application of liquid – liquid extraction includes:
a. Separating low concentration solutes and high boilers from aqueous solutions such as phenols.
b. Separating temperature sensitive compounds like acrylates and biotechnology.
c. Separating systems having similar boiling points such as aromatics from aliphatic hydrocarbons.
d. Separating mixtures with high boiling points like vitamins.
e. Metals extraction from wastewater. An example is copper.
f. Metals extraction from low grade ores.
g. Salt extraction from polymer solutions E.g. ketones, polyols and resins.
h. Organic compound extraction from salt solutions. Example is caprolactam
i. Azeotropic mixture separation by extracting formic acid from aqueous media using MTBE as solvent
j. Nuclear fuel recovery from purex process (Muller et al., 2015)
3. Advantages of Solvent Extraction
a. Solvent extraction has a wide range of application as the number of its unlimited practical combination can be achieved by varying the composition of the nature of binding agents or complexing agents and the composition of the organic phase.
b. Solvent extraction results in highly efficient rapid and very selective separation.
c. Solvent extraction can be carried out using simple equipment which could be automated.
d. Solvent extraction can be applied to a wide range of concentrations as its partition coefficients are usually independent of concentration.
e. Solvent extraction solution in some cases could be used in subsequent procedures directly. It can also follow back-extraction into aqueous solvents (Marlap, 2004).
4. Disadvantages of Solvent Extraction
a. If reaching equilibrium is very slow solvent extraction can be time consuming.
b. Small impurities in the solvent of solvent extraction can affect it.
c. For large number of samples, the process could be cumbersome.
d. When emulsion is formed this could interfere with the phase-separation process.
e. A minimum of 0.5 – 1.0ml is required by relatively large sample volumes.
f. Solvent extraction may require flammable or toxic solvents.
g. It uses organic solvents which is expensive and can generate organic wastes that is large in volume.
h. Time could be increased as solvent extraction may require multiple extraction which can also consume more materials and generate more wastes.
i. The effectiveness of the extraction and the distribution coefficient going from one phase to the other could be altered if the chemical form is changed.
j. The use of counter current process could be complicated and also require a complicated equipment. (Marlap, 2004).
C. Complex Ion Formation
When a metal ion or atom bonds with anions or molecules through an atom that can donate one or more electron pairs a complex is formed. A molecule or ion that can donate one electron pair is referred to a ligand. A compound which contains a complex ion is referred to as a coordination compound. Basically, there are no free ions in solution as every ion in the solution is surrounded by molecules of the solvent; hence every ion in a solution is complexed. Aquo ions are formed by complexed water molecules also known as inner hydration in aqueous solutions and the aquo ions may form a strong or weak bond (Marlap, 2004; Rodrigues et al., 2015).
An example of aquo ion is as follows:

- Classification of Ligands
The number of electron which a ligand can donate to a metal to formation of a coordination bond is a method used for the classification of ligands. The ligand is called a unidentate ligand if only one of its atom bonds with the metal. This is a category showing number of atoms with an electron pair which a ligand can donate in a complex ion -formation. In other cases where two, three, four, five or six atoms bond with the metal the ligands are called bidentate, tridentate, tetradentate, pentadentate or hexadentate respectively. The unidentate ligands are categorized by the number of electron pair available for donation which is one pair of electron while the bidentate ligands donate two pairs of electrons. Table 2.5 shows some common types of ligand (Marlap, 2004; Francisco et al., 2014; Rodrigues et al., 2015).
The term coordination number is used to represent the atoms which donates electrons to the metal atom. For instance, Ethylenediaminetetraacetate (EDTA) bonds to the metal in the example through two nitrogen atoms and four oxygen atoms. Table XX is a representation of ligands based on their types (Marlap, 2004; Rodrigues et al., 2015).
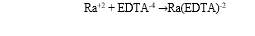
Furthermore, the basicity and nature of a ligand atom can be used to characterize the ligand as they can combine with any type of cation (metal ion) having a charge greater than one. Generally, compounds like Citrates, β -diketones, Acetates, and Tartrates complex all metals. However, the more selective complexing agents than oxygen donors are the sulfur donors, cyanide (CN-1), nitrogen donors and the heavy halides but they do not complex any of the A-metals in the periodic table. The cations of the transition metals and B-metals coordinate to sulfur, bromine, nitrogen, carbon, iodine and chlorine (Marlap, 2004; Francisco et al., 2014; Rodrigues et al., 2015).
Table 2.1 Common Types of Ligands
|
Type of Ligand |
Examples |
|
Unidentate |
Water (H2O), hydroxide (OH-1), carbonmonoxide (CO), halides (X-1), ammonia (NH3), thiocyanate (SCN-1), nitrite (NO2-1 |
|
Bidentate |
Citrate, Oxalate, ethylene diamine. |
|
Tridentate |
1,3,5 triaminocyclohexane, diethylene triamine |
|
Polydentate |
Organophosphates like tributylphosphate (TBP); trioctylphosphinic oxide (TOPO); (octyl(phenyl)-N,N-diiso-butylcarbamoyl-methylphosphine oxide [CMPO]); quaternary amines (tricaprylyl-methylammonium chloride [Aliquat-336]); tri-n-octylamin (TnOA); cryptates; macrocyclic polyethers (crown ethers like [18]-crown-6)
Ethelene diamine tetraacetic acid (EDTA), 8-hydroxyquinoline, diethylene triamine pentaacetic acid (DTPA), β-diketones (thenoylrifluoro acetone [TTA])
|
Adapted from: (Marlap, 2004)
2. Chelates
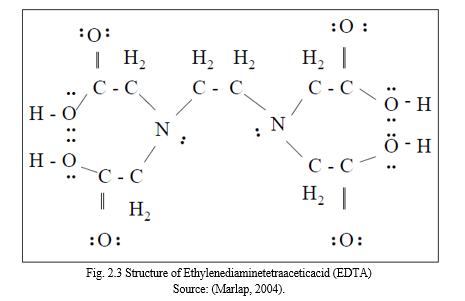
Chelates are multidentate ligands bound to ions or metal atoms by two or more electron pairs which forms ring structure. These multidentate ligands are also referred to as reagents or chelating agents. Basically, chelates are organic compounds which contains the carboxylic acid (RCOOH) amine (RNH2) functional groups in the order of two, four or six. When pH of the acid group is in an anionic form like in carboxylates (RCOO-1) the chelate is known to be effective but the lone pair of electrons of the nitrogen is not free for donating as it is not protonated. The bonds that chelates form with metals occur through the lone pair of electrons of these groups as bi, tetra or hexadentate ligands which forms a coordination complex with the metal. The metal is wrapped up in a claw-like fashion as the binding takes place in multiple sites hence the name chelates which means claw. In practice, on coordinating with the metals all chelates form a five or six-membered rings. Complex compounds formed by unidentate reagents are not as stable as chelates but the stability is improved if the multiple ring system is formed with a single metal ion or atom. A typical example is EDTA (Ethylenediaminetetraaceticacid) which is a hexadentate ligand; it forms a stable complex with most metals since it has four donor pairs from oxygen and two donor pairs from nitrogen. EDTA is commonly used as a complexing agent in basic solutions. Fig. 2.5 shows a typical Structure of Ethylenediaminetetraaceticacid (EDTA). When the hydrogen ion (H+1) is lost due to the ionization that occurs in the four carboxylic groups a stronger complexing agent (EDTA-4) is formed. Other usefulness of EDTA is in water as it is used to remove Mg and Ca to make the water soft and it is also used in food processing as an additive to increase the shelf life of food. This is because EDTA combines with the transition metal ions that speed up or catalyses the decomposition of food (Marlap, 2004; Francisco et al., 2014; Rodrigues et al., 2015).
Specificity for separating ions by selective bonding occurs because of most chelating agents binding more readily to certain cations. The complex can be collected by precipitation since the complex is occasionally insoluble under the solvent conditions used. Adjusting the pH of the medium which varies the net charge of the functional group would result in partial control of the selectivity of the chelate. The size of claw formed by the molecular structure and the number of functional groups available for bonding is a means through which various chelates provide specificity; hence a select fit for the diameter of a specific cation is provided. When coordination bond occurs, a ring system is formed with the metal atom by the electron-donating atoms of the chelate. Most times, complexes which are much more stable than unidentate ligands are formed by chelates. An example of this is the complex ion which is formed between bidentate ligand ethylenediamine (H2N-CH2-CH2-NH2, or en) and Ni+2, Ni(en)3+2 is almost 108 times more stable formed between ammonia and the metal ion. Ni(NH3)+2 (Marlap, 2004; Francisco et al., 2014; Rodrigues et al., 2015).
D. Diluent in Solvent Extraction
Diluent is a major portion of the extracting phase or may be a single liquid which does not extract the main solute. A diluent is a liquid which forms the solvent phase in solvent extraction and the modifiers and extractants are dissolved in it. A diluent is not referred to as a solvent but in some situation, it is referred to as a co-solvent. The research by Mowafy & Aly, 2001 revealed that both aromatic and aliphatic diluents were tested for the extraction of Th(IV) and U(VI) from 3M of HNO3 using a constant molar concentration of N,N,N',N'-tetrabutylsuccinamide (TBSA) (Mowafy & Aly, 2001)
- Coalescence, Dispersion and Mass Transfer
Drops move randomly in a turbulent dispersion and they collide with each other continuously. Between the collided drops is the intervening liquid which must drain to the thickness of the film rupture to allow the occurrence of agglomeration. The turbulent fluctuation could separate the droplets during drainage. The interaction force that exists between two droplets close to each are significant. However, it is not every collision that ends in coalescence so the efficiency of coalescence frequency in the drops of collision can be calculated as follows: Coalescence frequency = collision frequency X efficiency of coalescence. Dispersions that are physically equilibrated used for the prediction of distribution of droplet size, coalescence frequencies and average drop size can be well modelled by population balance equation. Yasuda et al., 2015 conducted a research to determine the effect of ultrasound on emulsification and demulsification. Irradiation of ultrasound sequentially at 20KHz and 4.8MHz greatly reduced the operation time needed to extract gallium for aqueous solution. Droplets are seen to scatter ultrasound and a secondary acoustic force is generated due to the interaction between the ultrasound and the droplets. Irradiated ultrasound at frequencies lower than 100 KHz into oil-water two-phase liquid causes the formation of emulsion and this is due to ultrasonic cavitation. The Fig. 2.6 below shows the complex interaction taking place in the extraction column (Kamp & Kraume, 2014; Yasuda et al., 2015; Kamp et al., 2016; Hasan, 2017).
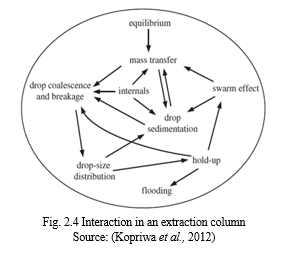
In the extraction column, different phenomena take place; for instance, drop coalescence, breakage and convection causes the changes seen in the drop-size distribution in an extraction column. There is a reduced volume -specific interfacial area with large drops while the small drops have mass transfer coefficients that is small and this is due to comparable rigid interface which hinders transporting transfer interface across the interface (Kopriwa et al., 2012; Kamp & Kraume, 2014; Kamp et al., 2016; Hasan, 2017).
A relatively narrow drop-size distribution would improve mass transfer. However, breakage and coalescence cause mixing inside the drops which also improves the transport of the transfer component into the drop from the surface layer; this has a positive effect on mass transfer. If the mass transfer or coalescence is relatively slow then the mixer-settler process can be used but the use of extraction columns would benefit from the higher volume-specific throughput. However, in an extraction column, the coalescence behaviour is determined in cell that are relatively small but a tedious evaluation of the changes that occur in drop-size distribution along a short column is required (Kopriwa et al., 2012; Kamp & Kraume, 2014; Kamp et al., 2016; Hasan, 2017).
E. Solid-Phase Extraction (SPE)
In the 1970s, solid-phase extraction (SPE) was introduced and it minimizes some of the disadvantages of liquid-liquid extraction (LLE). SPE is a solvent-extraction system with a stationary phase by adsorption onto a solid support (usually silica) and the other liquid phase which is mobile. It uses small membranes or columns and many of the extracting agents used in liquid-liquid extraction are also used in solid-phase extraction. SPE generates less amount of wastes and it is an excellent substitute for LLE as it is faster and more efficient. It requires samples as small as 50 – 500ul and small volumes of solvent not as pure as may be required by LLE. The working principles of SPE are like that of LLE were partitioning takes place between two immiscible liquids but in SPE the analytes to be extracted are partitioned between liquid and solid. The analyte needs to have a strong affinity for the solid phase than it has for the sample matrix. The compounds which is retained on the solid phase is later removed by eluting with solvents with very strong affinity for the analytes (Berrueta et al.,1995; Poole, 2000; Marlap, 2004; Zhang, 2013).
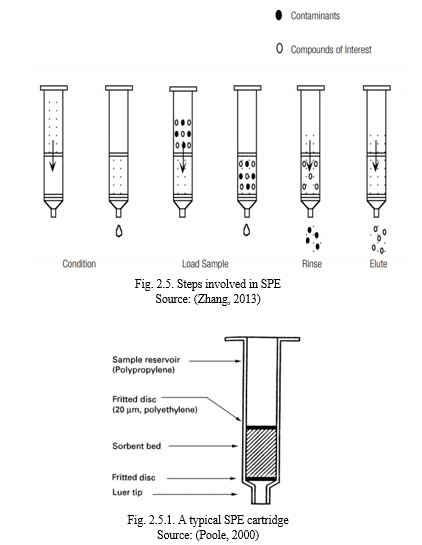
Intermolecular forces between the active sites on the adsorbent surface, analytes, liquid phase or matrix is the reason for the different mechanisms of retention or elution. In the procedure used for SPE, the liquid is passed through a polypropylene cartridge packed with adsorbent between two fritted disks. Fig. 2.5. and Fig. 2.5.1. shows the steps involved in SPE and a typical SPE cartridge. The liquid phase passes through the cartridge either by suction or by positive pressure like centrifuge, gravity or gas pressure. The SPE procedure can be categorized into five steps:
- Sorbent activation: The sorbent is activated by passing an appropriate solvent through to condition the solid’s surface.
- Removal of the activation solvent: a liquid similar in composition to the sample matrix is used to remove the activation solvent.
- Sample application: as the sample is passed through the sorbent retains the analyte. This is also called retention step or sorption.
- Washing step: a solvent which does not remove the analyte is used to remove any interfering compounds left in the previous step.
- Analyte elution: an appropriate solvent is used to elute the analytes from the adsorbent and collected for further analysis.
(Berrueta et al.,1995; Poole, 2000; Marlap, 2004; Zhang, 2013).
For inorganic oxides, silica gel, florisil, alumina and diatomaceous earth are the most important adsorbents for extraction and simplification of matrix. The most widely used general purpose adsorbent is silica gel made from sodium silicate the sol-gel procedure (Poole, 2000).
- Application of Solid-Phase Extraction
a. Adsorbent for Inorganic Oxides.
- Low and medium polarity analytes isolation from non – aqueous solutions
- Simplification of matrix through fractionating similar number and type of functional groups into group.
- Anions (alumina) and cations (alumina and silica) isolation from buffered aqueous solutions.
b. Low specificity sorbents for aqueous solution
- Ionizable and neutral analyte isolation from aqueous solution. (Weak bases and acids by ion suppression).
- Increase in solute size increases retention while ionization and polar interaction (hydrogen bonding) reduces retention.
- Weak retention is provided by polar bonded phases. This is useful if elution of analyte is a problem from non-polar sorbent.
c. Low specificity sorbents for organic solvents
- The size of solute is not important for retention but the type and number of functional group determines the retention.
d. Sorbent for ion exchange
- Ion exchangers which are strong are used to isolate the weak bases and weak acids of opposite charge and weak ion exchangers strong acid/base.
- Manipulating the sample ionic strength and pH gives control of the retention selectivity.
- Competing ion choice: selectivity is controlled by eluent pH and concentration.
(Berrueta et al.,1995; Poole, 2000; Marlap, 2004; Zhang, 2013).
2. Advantages of Solid-Phase Extraction Media
- Extraction with column/filter is very selective.
- Extraction with column/filter may not be attended to.
- The equipment required is not expensive.
- Liquid-liquid extraction can be correlated with solid-phase extraction.
- Low volume of waste is generated.
- SPE can be applied to samples dissolved in media that is very acidic.
3. Disadvantages of Solid-Phase Extraction Media
- The flow rate of 1-3 mL/min through column is slow.
- Cost is a factor in solid-phase extraction as the extraction columns cannot be reused.
- Contaminants such as suspended matter may be filtered by media and carried into the next separation step.
(Berrueta et al.,1995; Poole, 2000; Marlap, 2004; Zhang, 2013).
Conclusion
This paper summarized the processes involved in solvent extraction. |The distribution coefficient and partition coefficient are used to quantitatively determine the degree of solubility of a solute in a solvent compared to its solubility in another solvent. The solvent used in LLE must have high affinity for the solute to be extracted and it must not be completely miscible with the carrier liquid. Many of the extracting agents used in LLE are also used in SPE. SPE generates less amount of wastes and it is an excellent substitute for LLE as it is faster and more efficient. SPE requires samples as small as 50 – 500ul and relatively small volumes of solvents not as pure as may be required by LLE. More research would be needed in the area of diluents in solvents extraction and also use of ionic liquids for selective separation in solvent extraction.
References
[1] Baba, Y., Kubota, F., Kamiya, N., Goto, M., 2011. Recent advances in extraction and separation of rare earth metals using ionic liquids. Journal of Chemical Engineering. 44(10): 679–685. [2] Berrueta, L. A., Gallo, B., Vicente, F., 1995. A Review of Solid Phase Extraction: Basic Principles and New Developments. Chromatographia. 40: (7-8): 0474-0475. [3] Chizhkov, V. P., Boitsov, V. N., Demin, A. V., 2011. General Separation Theory and Liquid Extraction. Russian Journal of Physical Chemistry A. 85(3): 494–498. [4] Demim S., Drouiche N., Aouabed A., Benayad T., Dendene-Badache O., Semsari S., 2013a. Cadmium and nickel: Assessment of the Physiological Effects and Heavy Metal Removal using a Response Surface approach by L. Gibba. Ecological Engineering. 61: 426–435. [5] Demim S., Drouiche N., Aouabed A., Semsari S., 2013b. CCD Study on The Ecophysiological Effects of Heavy Metals on Lemna Gibba. Ecological Engineering. 57: 302–313. [6] Demim S., Drouiche N., Aouabed A., Benayad T., Couderchet M., Semsari S., 2014. Study of Heavy Metal Removal from Heavy Metal Mixture Using the Ccd Method. Journal of Industrial & Engineering Chemistry. 20: 512–520. [7] Francisco, V., Basilio, N., Garcia-Rio, L., 2014. Ionic Exchange in p-Sulfonatocalix[4]arene-Mediated Formation of Metal Ligand Complexes. Journal of Physical Chemistry B. 118 (17). [8] GBH, no date. Liquid – Liquid Extraction: Basic Principles. GBH Enterprises. Process Engineering Guide. [9] Hamburg G., 2017, Thermal Process Engineering: Liquid-liquid extraction and solid-liquid extraction. Available at: http://www.gunt.de/images/download/extraction_english.pdf. [10] Hasan, B.O., 2017. Breakage of drops and bubbles in a stirred tank: A review of experimental studies. Chinese Journal of Chemical Engineering. 25: 698–711 [11] Kamp, J. & Kraume, M., 2014. Influence of drop size and superimposed mass transfer on coalescence in liquid/liquid dispersions – Test cell design for single drop investigations. Chemical Engineering Research and Design. 92: 635–643 [12] Kamp, J., Hansch, R., Kendzierski, G., Kraume, M., Hellwich., O., 2016. Automated image analysis for trajectory determination of single drop collisions. Computers and Chemical Engineering. 89: 184–191. [13] Koch, J. and Shiveler, G. 2015. Design Principles for Liquid-Liquid Extraction. Alche Publication CEP Magazine. Available at: https://www.aiche.org/resources/publications/cep/2015/november/design-principles-liquid-liquid-extraction [Accessed: 24th May 2017]. [14] Kopriwa N., Buchbender F., Ayesterán J., Kalem K. & Pfennig A., 2012. A critical review of the application of Drop-population balan ces for the design of Solvent extraction columns: I. Concept of Solving drop-population balances and Modelling breakage and coalescence. Solvent Extraction and Ion Exchange. 30: 683–723. [15] Leyma R., Platzer S., Jirsa F., Kandiolla W., Krachler R., & Keppler B.K. 2016. Novel thiosalicylate-based ionic liquids for heavy metal extractions. Journal of Hazardous Materials. 314: 164 – 171 [16] Marlap, 2004. Separation Techniques. Multi-Agency Radiological Laboratory Analytical Protocols Manual, USEPA. 2: 14 – 231 [17] Mowafy E.A. & Aly H.F., 2001. Synthesis and characterization of N,N,N\',N\'-tetrabutylsuccinamide as extractant for uranium(VI) and thorium(IV) ions from nitric acid solution. Journal of Radioanalytical and Nuclear Chemistry. 250(1): 199–203. [18] Muller E., Frankurt A.M., Berger R., Blass E., Slyutts D., Bayer A.G., Pfennig A. & Aachen R., 2015. Liquid-Liquid Extraction. Encyclopedia of Industrial Chemistry. Ulmann’s Academy. 1-2. [19] Poole C.F., 2000. Solid -Phase Extraction. Academic Press - Extraction. 1405 – 1416 [20] Rodrigues, S.S.M., Prieto, D.R., Ribeiro, D.S.M., Barrado, E., Prior, J.A.V., Santos, J.L.M., 2015. Competitive metal–ligand binding between CdTe quantum dots and EDTA for free Ca2þ determination. Talanta. 134: 173–182. [21] Tarihi, G., 2001. The Distribution Coefficients of Acetic Acid between Water and Solvent Systems. Journal of Engineering Sciences.7(3): 415 – 419 [22] Xie F., Zhang T.A., Dreisinger D., Doyle F., 2014. A critical review on solvent extraction of rare earths from aqueous solutions. Minerals Engineering. 53: 10-28. [23] Yang F., Kubota F., Baba Y., Kamiya N. & Goto M., 2013. Selective extraction and recovery of rare earth metals from phosphor powders in waste fluorescent lamps using an ionic liquid system. Journal of Hazardous Materials. 254– 255: 79– 88. [24] Yasuda K., Nguyen T.T., Okura R., Nakayama S., Asakura Y. & Jin J., 2015. Dispersion and coalescence of oil droplets by ultrasound and application for solvent extraction of gallium. Japanese Journal of Applied Physics. 54: 1-6. [25] Zhang, A.J., 2013. Why Automated Solid-Phase Extraction Is Superior to Traditional Extraction. Thermo Scientific. 1-4.
Copyright
Copyright © 2023 Dr. Aghogho Blessing Obukohwo. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.

Download Paper
Paper Id : IJRASET48272
Publish Date : 2022-12-21
ISSN : 2321-9653
Publisher Name : IJRASET
DOI Link : Click Here
 Submit Paper Online
Submit Paper Online