

Ijraset Journal For Research in Applied Science and Engineering Technology
Role of in silico Drug Design in Pharmaceutical Sciences
Authors: Rainul Islam, Sumit Maji, Souparna Kabiraj, Umme Habiba, Rohan Pal, Somenath Bhattacharya, Soumallya Chakraborty, Dr. Arin Bhattacharjee
DOI Link: https://doi.org/10.22214/ijraset.2022.42836
Certificate: View Certificate
Abstract
In silico drug design is the study to identify, develop, analyze, optimize drugs or biologically cum pharmaceutically active compounds by using computerized software programs as well web servers. In silico drug design is commonly known as computer aided drug design or CADD in short. This technique shows a vital role in preclinical drug design and development. CADD can improve the speed of drug design. It reduces time as well as total cost of the experiments. Potent cum suitable molecules are prepared after performing in silico drug design including CADD. Various applications like confirmation generation, homology modeling, multiple sequence alignment, molecular docking study, generation of Pharmacophores, virtual screening, de novo drug design, QSAR (Quantitative structure activity relationships) study, molecular modeling, in silico ADMET (Absorption, distribution, metabolism, excretion and toxicity) prediction of CADD has been implemented to design newer molecules. The current study focuses on different strategies cum approaches through computer aided drug designing applied to find potent, efficient and safe molecules in the field of drug discovery.
Introduction
I. INTRODUCTION
Drug design & discovery is a process to find newer active, potent and safe molecules. In silico drug design is one of the processes under drug design through different computerized software and web servers [1]. It is very long process. It takes about many years to find newer molecules.
It is extremely expensive methods. Drug design is very important now a day as because most of the drugs are resistant. They are not activated in body when they are administered due to resistance. So those kinds of drugs are inactivated. To prevent the resistance, drug design is very necessary parameters in Pharmaceutical field. Drug design generally starts from identification of target to validation of newer compounds. This designing process is usually having 5-6 main steps. Different steps include identification of target, validation of target, lead identification, lead optimization and then comes to find potent compounds after its ADMET prediction and trial [1-2].
CADD is two types, one is structure based drug design, another one is ligand based drug design. Strategies like Homology modeling, confirmation generation of ligand and receptor, finding of active site of receptor, molecular docking, pharmacophore generation, QSAR modeling, ADMET prediction, statistical analysis have been used in this process [3-4]. Different types of software programs cum web servers like Chem sketch, Protein data bank (PDB), Chem draw, UCSF Chimera, Rasmol, auto dock tools, Pharmagist, ZINC, E-dragon, Padel descriptors, ALOGPS, etc. are used to design and find newer molecules [5-6]. Applications of CADD can spread over a larger area to find different potential inhibitors against different diseases. Many newer diseases came to the world and no drug of choice is available to treat the diseases. Newer molecules can be designed easily through CADD.
II. STEPS OF DRUG DESIGN AND DISCOVERY [1-2, 7]
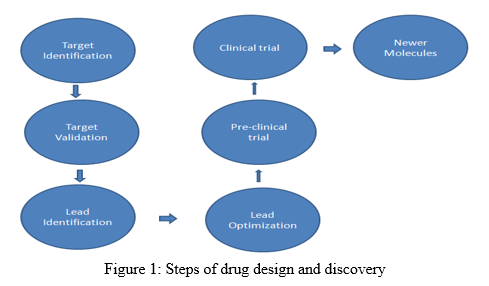
III. DIFFERENT APPROACHES OF CADD [1-5, 8]
- Bioinformatics including Homology modeling and prediction of active site of receptor
- Cheminformatics
- Molecular docking
- Pharmacophores generation
- Virtual screening
- De novo drug design
- QSAR
- Molecular modeling
- In silico ADMET prediction
A. Bioinformatics
Bioinformatics is the study to find and design receptor structure. It is the branch of science in which computer science, biological science and information technology merges in a single discipline [4, 7-9]. Different types of web servers like Protein data bank can be used to find the structure of receptors. Software programs and web servers like Modeller, Swiss prot, ClustalW, Rasmol, EMBOSS, ENTREZ, BLAST (Basic local alignment search tool), FASTA can be used to find unknown structures of receptors cum sequences [4, 9-10]. Homology modeling is the study to find the Crystallographic and complex structures of unknown receptor [4, 11]. At first, sequence can be noted, then preliminary structure of unknown receptor has been developed and it can be validated through software programs. After finding of receptor structures, active site of receptors can be identified through ligplot. Ligplot is the receptor-ligand interaction. Different web servers are available in where ligplot can be done [4, 12-14]. From ligplot, X,Y and Z coordinates of receptors can be noted. Bioinformatics is one of the major step of computer aided drug designing as because the importance of receptors study in respect of finding newer molecules [4, 15].
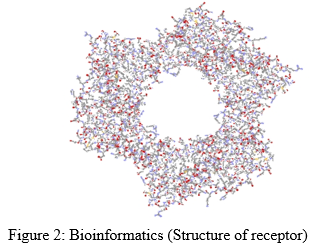
B. Cheminformatics
Cheminformatics is having its vital importance in CADD. Cheminformatics is the process to study the structure, properties of ligands. It is also called as chemical informatics. Without ligand structure, CADD study cannot be verified. Different types of software programs cum web servers can be used to find, draw and design newer molecules. By using Chem Sketch, Chem Draw, Marvin draw, Pubchem web server, Chemspider web server, etc [1-3, 16-20]. ligand structure can be prepared. Different properties of ligands can be checked by using different software programs. In QSAR, we can find the molecular descriptors of the ligand. The molecular descriptors are also called as physicochemical properties of the ligand. These physicochemical properties can also be checked through different servers like padel descriptors, ALOGPS, E-dragon web server, etc [1-3, 21].
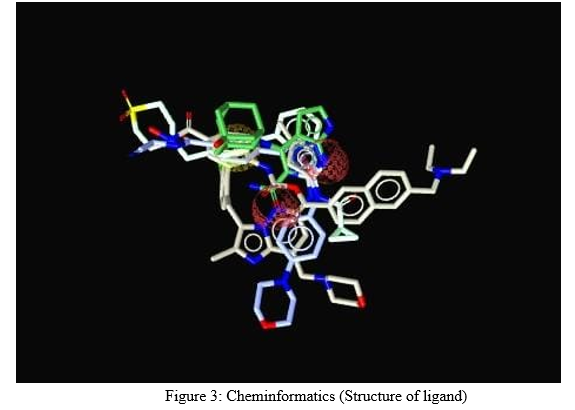
C. Molecular Docking
It is the process of receptor-ligand interactions. Receptor is a macromolecules or bigger shape in size. These receptors are having too many interaction sites. These are basically protein in nature. The ligand is a small molecule as compared to receptor. The receptor has one binding pocket or site. Basically, this site is suitable for receptor-ligand interactions [1-4, 22]. This site is called as active site of receptor. The ligand is fitted to the active site of receptor. The theory or principles of docking is based on lock and key mechanism in where lock as receptor and key as ligand. The attachment of ligand to the active site of receptor can be done by strong intermolecular interactions like hydrogen bond formation, ionic bond creation, etc. So in words, molecular docking is the study to observe the attachment of ligand to the active site of receptor through intermolecular interactions [1, 23-26]. Intermolecular interactions can be calculated by measuring docking score or binding energy. Different types of software programs can be used for molecular docking. Auto dock tool is one of the software used to measure binding energy. It is also used for docking analysis like validation of docking. This tool can be used to generate huge numbers of poses of ligand (molecule) in the binding site (active site) of the targeted receptor and also used to find affinity for binding of each pose of ligand [27]. Different package software programs can be used to find docking score of each pose of ligand. This type of software is basically worked force field under molecular mechanics. Force field is the total energy of each pose of a ligand to the active site of receptor. The software like auto dock tools is basically having one input point and output point. At first the conformation of ligand and receptor can be prepared. In input, the ligand and receptor conformer can be given with active site coordinates of the targeted receptor [28]. After given input data, different program of the software can be run. In the output part of the software, the binding affinity cum energy can be auto calculated with respect of too many pose of ligand to the active site of the receptor [29]. Docking analysis can be done after finding output data containing binding energy. Docking analysis is the process to check or validate the correctness of the docking of ligand to the active site of receptor. Different types of docking are available now a day’s like Rigid binding, Flexible binding, Semi-flexible docking, etc [28-30].
- Rigid Docking: In rigid docking, the receptors and ligands are rigid in nature. They are not flexible. Different criteria’s for ligand like bond length, bond angles, torsion angles, etc are not modified during docking. In this type of docking the molecule or ligand cannot change or alter their spatial shape. Patch dock is an application for molecular docking [1-3, 31]. The principle of the application is based on size and shape of complementary. Through this application, at first molecular shape representations like convex, concave, flat surface patches, etc can be done. After that by implementing surface patch matching techniques, convex patches are matched with concave and flat patches. Next step is filtering based on the successful rate of binding of ligand to the active site of receptor. Therefore, the unacceptable docking is not accepted due to steric hindrance. The highest fitted ligands to the active site of receptors can be selected. Another one program is FRED (Fast rigid exhaustive docking) [1-5, 32].
- Flexible Docking: Flexible docking is the process in where input molecules are flexible or the receptors and ligands are flexible in nature. The receptors and ligands can flexibly interact with each other and they are able to form conformer. Strong intermolecular interactions created due to flexible attachment of both. Flex dock is one of the applications under flexible docking [1-4, 33].
- Semi-Flexible Docking: In case of semi-flexible docking, the receptor is basically rigid or fixed and the ligand is flexible [1-3, 34].
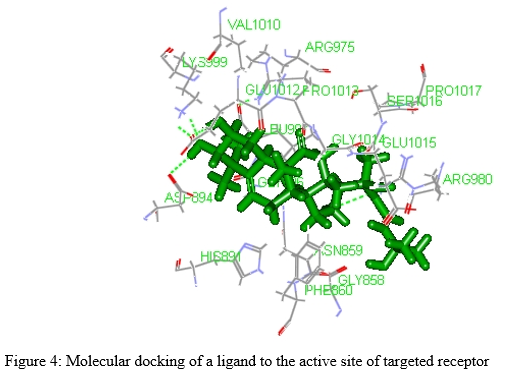
Molecular docking is an important application of computer aided drug design. Newer lead molecules can be obtained by lead identification and optimization techniques.[by molecular docking]
D. Pharmacophores Generation
Pharmacophore is the features of molecules that are very important to show biological activity for molecules. Based on the molecular features, the molecules can provide the biological activity. The molecules shows the biological activity only for its pharmacophoric features. Various types of software programs, web servers like Pharmagist, Ligandscout, CATALYST, etc can give the pharmacophoric features of molecules [1-5, 22, 35]. Different types of pharmacophoric features are [1-5, 22, 36]:
- Hydrogen Bond Acceptor: This type of feature usually fit with sp or sp2 nitrogen atoms, sp or sp3 oxygen or sulphur atoms include lone pair and charge less than or equal to zero.
- Hydrogen Bond Donor: This type of feature usually fit thiols, acetylenic hydrogens, non-acidic hydroxyls groups.
- Hydrophobic: This type of feature usually fit phenyl, cycloalkyl, isopropyl and methyl groups.
- Negative Ionisable: This type of feature usually fit Trifluoromethyl sulphonamide hydrogens, sulphonic acids, phosphonic acids, tetrazoles.
- Positive Ionisable: This type of feature usually fit basic amines, basic secondary amidines.
- Ring Aromatic: This type of feature usually fit 5 or 6 membered aromatic rings.
E. Virtual Screening
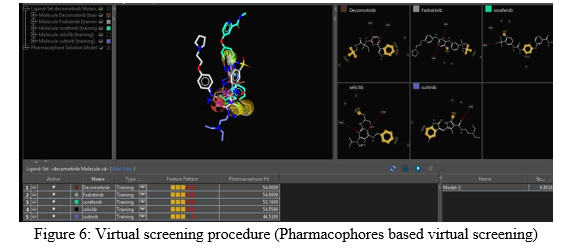
Virtual screening is the screening process of larger database into smaller database. This technique is also called as VS in short. It is a searching tool by that way similar types of ligands bind to the active site of receptor can be identified from the tool. In this techniques, large set of libraries include molecules database can be identified based on pharmacophoric features, receptor-ligand interactions, etc [35-37]. The molecules are screened and filtered from larger set database of molecules. Screening can also be done by filtering different pharmacokinetic (Absorption, distribution, metabolism and excretion) and toxicity parameters. It is one of best approach for virtual screening technique. By this technique, maximum molecules are filtered. Different types of software programs, web servers like ZINC, ligandscout, etc can be used for virtual screening. Virtual screening is basically three types [35, 38].
- Ligand Based Virtual Screening: In this screening, pharmacophoric features of known molecule can be identified. Similar molecules based on size, shape, etc from the pharmacophoric features search through virtual screening tools and matched with known molecules to find best interaction in between receptor-ligand [35, 39].
- Structure Based Virtual Screening: In this method, based on receptor-ligand interactions (docking score) screening can be done. The goal of the method is to construct unknown protein structure (homology modelling) and find protein interaction sites. According to the concept, active molecules can be screen through virtual screening software programs cum web servers from large libraries of molecules. Through molecular dynamic simulations, virtual screening can also be possible [35, 40].
F. De Novo Drug Design
De novo drug design is one of vital applications of CADD. This technique is used to find structures of new lead molecules for fitting into the binding site of receptor. Different types of docking software programs, web servers can be used for de novo drug design. There are two types of de novo drug design [1-3, 41].
- Template Process: The template structure can be identified and modified through template method. This template structure should be fitted to the active site of the targeted receptor to develop binding affinity. This type of method basically starts from searching or identifying the database for suitable template structures. Software for an example like CAVEAT can be used to identify the template structure and fit this configuration to the active site of receptor to improve its binding energy [1-5, 42].
- Component Fragment Process: The binding site of receptor is having different atoms and groups. These atoms and groups can create intermolecular interactions like hydrogen bonding with a suitable molecule or ligand. Software program like LUDI can identify and select the fragments based on the size cum shape of binding site of targeted receptor. This program also fit these fragments to the binding site of receptor to check, improve and validate the binding affinity. In this method, all of the structures are rigid in nature and the intermolecular interactions like bonds can show degree of free rotation [1-3, 43].
G. QSAR
QSAR is Quantitative structure activity relationship. QSAR study is very important in CADD. The main aim or objective of QSAR is to find the molecular descriptors cum physiochemical properties of compounds and correlates the physiochemical parameters to the biological activity of compounds [1, 44]. This is a statistical method. Another one objective of QSAR is to improve potency and efficiency of compounds. QSAR study is used to validate the compounds [1-2, 45]. This study saves cost of practical experiments. QSAR supports green chemistry. Molecular descriptors are the basic features cum subparts of the compound structure. Through different software programs and web servers, molecular descriptors of compounds can be calculated. After calculating molecular descriptors of compounds, statistical and mathematical applications through graphical representations (physicochemical parameters vs biological activity) can be implemented to improve the efficiency cum potency of the compounds. If all the data of physicochemical parameters and biological activity of compounds are suitably explained or correlated through different statistical analysis then the aim cum objectives of QSAR study is successful [9, 46]. Molecular descriptors are divided into three divisions like biological properties, intrinsic properties and physicochemical properties. Examples of biological properties are LD50, IC50, MIC (Minimum inhibitory concentration), etc. Intrinsic properties are linked to the structure of compound. Intrinsic properties are molecular weight, molecular volume, surface area, etc. Physicochemical properties are divided into three subparts like partition parameters or hydrophobicity parameters, steric parameters and electronic parameters [22, 47]. Hydrophobicity parameters describes that how the compounds can quickly cross the cell membrane. These parameters improve the receptor-ligand intermolecular interactions. By changing different substituents of the compound, hydrophobicity of compounds can be changed [22, 48].
- Partition Effects: Partition parameters or hydrophobicity parameters are partition coefficient, lipophilic substitution coefficient, distribution coefficient, etc. Partition coefficient (P) is defined as the ratio of unionized drugs between two liquid (n-octanol and water) phases.
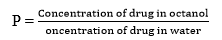
logP is defined as the logarithmic ratio of concentration of the unionized drug in two phases. Hydrophobic molecules prefer generally high partition coefficient value as because they can able to dissolve in n-octanol phase or layer. Hydrophilic molecules prefer generally low partition coefficient value as because they can able to dissolve in water or aqueous phase or layer. Distribution coefficient (D) is the ratio of concentration of unionized and ionized drugs in between aqueous medium and organic solvent [22, 49].
2. Electronic Effects: Electronic parameters of different substituents of compounds are used to measure electron donating or electron withdrawing abilities of substituents. Electronic factors of substituent of a compound may create the affect on compound’s polarity or ionization. One example of electronic parameter is Hammett substituent constant. Hammett constant is the ability of electron donates or electron withdraws of a substituent of compound [1-3, 50]. This value can be calculated by determining dissociation of substituted benzoic acid and dissociation of parent benzoic acid (without substitution) [1-5, 51]. After that those values for substituted compound and parent compound can be compared. Benzoic acid is weak acid therefore it can dissociate or ionizes in water moderately. If the substituent of parent benzoic acid is having the ability to withdraw electron then entire aromatic ring with carboxylate anion after dissociation can be more stable. On the other hand, parent benzoic acid with substituents is having the capacity to donate electron then entire aromatic ring with carboxylate anion after dissociation can be less stable compared to previous one. Hammett substituent constant is not applicable for the substitution on ortho position as because in ortho position, the substitution can show the steric effect. This constant is suitable for the substation on meta and para position [2-5, 52].
3. Steric Effects: Steric parameters or factors represent that how the size and shape of a drug can correlate with the binding affinity cum interaction to the active site of the receptor. Steric parameters are Taft’s steric factors, verloop steric factors, molar refractivity, etc [1-3, 53-54]. Steric factors are also related to the bulkiness of the compound. The bulky substituent of a compound generates strong intermolecular interactions in between the molecule and receptor. So the binding affinity can be increased. Taft’s steric factor can be measured by finding the hydrolysis rate of substituted aliphatic ester and comparing it with a standard parent ester in acidic conditions. Another one steric factor is verloop steric parameter. These parameters are bond angles, bond length, van der waal radii, etc. Basically, the steric parameters are calculated for the substituted compounds through computerized software program sterimol [1-3, 55-56]. Molar refractivity is one of the steric parameter. It is expressed by MR. Molar refractivity is the measurement of volume captured by atoms. MR is calculated by this equation [1-3, 57-58]:

Therefore, the physiochemical properties of compound can be defined as the physical properties, chemical properties, and structural properties cum parameters.
QSAR study is divided in several categories like 2D-QSAR (Hansch analysis, Free-wilson approach), 3D-QSAR (COMFA, COMSIA), etc.
a. Hansch Analysis
Hansch proposed that the biological activities of drugs are correlated with the combination of physicochemical properties or parameters of drugs. This incident can be explained through Hansch equation [1-3, 59-60].

Different types of physicochemical properties like partition coefficient, Hammett substituent constant, Taft’s steric factor, Verloop steric factor, Molar refractivity, etc can be measured by using different descriptors finding software programs like padel descriptors, ALOGPS, E-dragon, etc. Then the graphical plot of biological activity vs physicochemical properties of drug can be drawn and after that statistical analysis can be performed. If all the data are successfully correlates with each another then QSAR model is validated. If regression value is above 0.9 then the QSAR study is successfully completed.
b. Free Wilson Approach
In this method at first biological activity of parent molecule is identified. The substitution on parent molecule can be done. Then the biological activity of substituted compound can be calculated. The biological activity of parent molecule is compared with the activities of substituted analogues. This approach expressed by one equation [1, 61]:
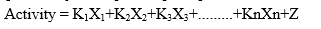
Where, Xn = Indicator variable (value 1 or 0)
Kn = The contribution that each substituent show the activity
Z = Constant showing the overall average activity of all the structure
This method is very critical for rationalizing the results cum data. This method cannot able to describe that why a substituent on the compound at a particular position is good, bad or best for showing the biological activity.
c. 3D-QSAR (COMFA, COMSIA)
In 3D-QSAR, different variables of electrostatic and steric interactions like hydrophobic, hydrogen bond acceptor, hydrogen bond donor, etc of molecules are identified through different descriptor generating software programs. Through PLS (Partial least square) statistical study is used to plot variables cum different molecular descriptors against biological activity to correlate the biological activity with molecular descriptor variables. CoMFA is comparative molecular field analysis and CoMSIA is comparative molecular similarity indices analysis. 3D-QSAR technique is most advanced technique as compared to 2D-QSAR [2-3, 62].
H. Molecular Modeling
Molecular modeling is the study of designing, representing, identifying, modifying, and generating the 3D-structures of compounds with its different physical and chemical properties. The objective of molecular modeling is to improve the biological activity of the compound through generating intermolecular interactions in between receptor and ligand. Different types of computerized tools are used to find the conformations of molecules by predicting vibrational conformations. Intermolecular interactions can be measured by finding total energy of the molecule. Total energy is basically the combinations of two types of interactions like bonded and non bonded interactions whereas bonded interactions include bond angle stretching, bond angle bending, rotation of torsion energy, bond angle, etc as well as non bonded interactions include van der waal, coulomb interactions, etc [48, 63]. Energy minimization is very important technique under molecular modeling. Sometimes unwanted intermolecular interactions can be present. Energy minimization is used to modify these interactions and calculate total energy of the molecule. There are two types of molecular modeling such as molecular mechanics and quantum mechanics [64].
- Molecular Mechanics: Molecular mechanics describes the number of atoms and how the study of atoms can influence the molecular behaviour. Molecular dynamic study is one of the important approach under the molecular mechanics study in where this study can be used to find the motion including kinetic energy of molecules.
- Quantum Mechanics: Quantum mechanics is used to generate quantized energy. The main goal of the study is to the find the behavioural characterization of electron and nuclei as well as orbitals of atoms.
I. In silico ADMET Prediction
This step is very important to check pharmacokinetic properties of drugs like ADME (Absorption, distribution, metabolism and excretion) and toxicity. This step is carried out after all of the important functions regarding drug design in CADD. Most of the compounds are screened through this technique. Several software programs like TOPKAT, DEREK, Multi CASE tool, etc can able to find the ADMET of drugs. This step is based on the Lipinski’s rule of filtration. Some of important parameters of the molecules associated with Lipinski’s rule can be checked through different software programs cum web servers. Lipinski’s rule was discovered by Christopher A. Lipinski. The rule explained that orally active drugs must main following criteria [65]:
- Hydrogen bond donor (HBD) in a compound is not greater than 5.
- Hydrogen bond acceptor (HBA) in a compound is not greater than 10.
- Molecular weight (MW) of a compound is less than 800 gms or 500 daltons.
- Partition coefficient (logP) of a compound is not more than 5.
- The range of polar surface area (PSA) of a compound is not more than 190 A2.
- Molar refractivity (MR) range of a compound is within 40-130.
- The number of total atoms in a compound is within 20-70.
- The number of total rotatable bonds in a compound is not more than 10.
Conclusion
Computer aided drug design or CADD in short is very important approach under in silico drug design. CADD is divided in two types like ligand and structure based CADD. These two methods are very useful to find and design potent, efficient, active newer safe molecules. These methods save the time as well as cost of drug design process. Now a day’s different types of new diseases came in front of us. Besides, drug resistance is a huge problem for treating existing diseases as because too many drugs are resistant in human body. Due to this resistance, the existing drugs are not able to fight against the diseases. So it’s a challenge to design and validate newer molecules quickly to treat the diseases. Through in silico drug design include CADD, we can rapidly design, identify and validate newer potent safe molecules against the diseases. Conflict of Interest: Nil
References
[1] Hoque I., Chatterjee A., Bhattacharya S. and Biswas R.; An approach of computer-aided drug design (CADD) tools for in silico pharmaceutical drug design and development; International Journal of Advanced Research in Biological Sciences; 2017; 4(2); 60-71. [2] Anderson A.C.; The process of structure-based drug design; Chemistry & Biology; 2003; 10(9); 787-797. [3] Veselovsk A.V. and Ivanov A.S.; Strategy of computer-aided drug design; Russian Academy of Medical Science; 2003; 3(1); 33-40. [4] Vyas V.K., Ukawala R.D., Ghate M. and Chintha C.; Homology modeling a fast tool for drug discovery: current perspectives; Indian Journal of Pharmaceutical Sciences; 2012; 74(1); 1-17. [5] Talele T.T., Khedkar S.A. and Rigby A.C.; Successful applications of computer aided drug discovery: moving drugs from concept to the clinic; Current Topics in Medicinal Chemistry; 2010; 10(1); 127-141. [6] Zhou S.F. and Zhong W.Z.; Drug design and discovery: principles and applications; Molecules; 2017; 22(2); 279. [7] Dutta S. and Sachan K.; Computer-aided drug design a new approach in drug design and discovery; International Journal of Pharmaceutical Sciences Review and Research; 2010; 4(3); 146-151. [8] Gilda H.L., Hugo O.V. and Ibon A.; Strategies for indirect computer aided drug design; Pharmaceutical Research; 2013; 10(4); 475- 486. [9] Denis F., Eugene M. and Alexander T.; Trust but verify: On the importance of chemical structure curation in cheminformatics and QSAR modeling research; Journal of Chemical Information and Modeling; 2010; 50(7); 1189–1204. [10] Hopfinger A.J.; Computer-assisted drug design; Journal of Medicinal Chemistry; 1985; 28(9); 1133-1139. [11] Gaba M., Gaba P., Singh S. and Gupta G.D.; An overview on molecular docking; International Journal of Drug Development and Research; 2010; 2(2); 219-231. [12] Vijayakrishnan R.; Structure-based drug design and modern medicine; Journal of Postgraduate Medicine; 2012; 55(4); 301-304. [13] Daina A., Blatter MC., Baillie G.V., Palagi PM., Marek D. and Xenarios I.; Drug design workshop: a web-based educational tool to introduce computer-aided drug design to the general public; Journal of Chemical Education; 2017; 94(3); 335-344. [14] Bisht N. and Singh B.K.; Role of computer aided drug design in drug development and drug and development, International Journal of Pharmaceutical Sciences and Research; 2018; 9(4); 1405-1415. [15] Scotti L. and Scotti M.T.; Computer aided drug design studies in the discovery of secondary metabolites targeted against age-related neurodegenerative diseases; Curr. Top. Med. Chem.; 2015; 15(21); 2239–2252. [16] Baig M.H., Ahmad K., Roy S., Ashraf J.M., Adil M., Siddiqui M.H., Khan S., Kamal M.A., Provazník I. and Choi I.; Computer aided drug design: success and limitations; Curr. Pharm. Des.; 2016; 22(5); 572–581. [17] Dharampreet S., Ramanjeet K., Patil R.K. and Patil H.C.; A review on: computer aided drug design, International Journal of Emerging Technologies and Innovative Research; 2020; 7(3); 1165-1176. [18] Kapetanovic I.M.; Computer-aided drug discovery and development (CADDD): in silico-chemicobiological approach.; Chem. Biol. Interact.; 2008; 171(2); 165–176. [19] Shekhar C.; In silico pharmacology: computer-aided methods could transform drug development; Chemistry & Biology; 2008; 15(5); 413-414. [20] MartiRenom M.A., Stuart A.C., Fiser A., Sanchez R., Melo F. and Sali A.; Comparative protein structure modelling of genes and genomes; Annual Review of Biophysics and Biomolecular Structure; 2000; 29(1); 291-325. [21] Pfisterer J.H., Liang Y., Schneider O. and Bandarenka A.S.; Direct instrumental identification of catalytically active surface sites; Nature; 2017; 549(7670); 74-77. [22] Udugade B.V. and Gawade S.P.; 3D QSAR and pharmacophore modeling on substituted cyanopyrrolidines as type ii anti-diabetic agents potential dipeptidyl peptidase-iv inhibitors; Pharmacophore (An International Research Journal); 2016; 7(5); 342-348. [23] Mahdi M., Szojka Z., Mótyán J.A. and Tozsér J.; Inhibition profiling of retroviral protease inhibitors using an HIV-2 modular system; Viruses; 2015; 7(12); 6152-6162. [24] Coussa R.G. and Kapusta M.A.; Treatment of cystic cavities in X-linked juvenile retinoschisis: The first sequential cross-over treatment regimen with dorzolamide; American Journal of Ophthalmology Case Reports; 2017; 1(8); 1-3. [25] Gradman A.H., Schmieder R.E., Lins R.L., Nussberger J., Chiang Y. and Bedigian M.P.; Aliskiren, a novel orally effective renin inhibitor, provides dose-dependent anti-hypertensive efficacy and placebo-like tolerability in hypertensive patients; Circulation; 2015; 111(8); 1012-1018. [26] Jadhav M., Yeola C., Zope G. and Nabar A.; Aliskiren, the first direct renin inhibitor for treatment of hypertension: the path of its development; Journal of Postgraduate Medicine; 2012; 58(1); 32. [27] Das S. and Vardhan A.; Computer aided drug designing; Int. J. Med. and Dent. Sci.; 2017; 6(1); 1433-1437. [28] Tang Y., Zhu W., Chen K. and Jiang H.; New technologies in computer-aided drug design: toward target identification and new chemical entity discovery; Drug Discovery Today: Technologies; 2006; 3(3); 307-313. [29] Kumar S.C.; An insight to drug designing by in silico approach in biomedical research; J. Pub. Health Med. Res.; 2013; 1(2); 63-65. [30] Kola I. and Landis J.; Can the pharmaceutical industry reduce attrition rates?; Nature Reviews Drug Discovery; 2014; 3(8); 711-716. [31] Mishra S.S., Sharma C.S., Singh H.P., Kumar N. and Pandiya H.; In silico pharmacokinetic and toxicity study of some selected antidepressant drugs; Chemistry Research Journal; 2017; 2(1); 42-45. [32] Rahman M.M., Karim M.R., Ahsan M.Q., Khalipha A.B.R., Chowdhury M.R. and Saifuzzaman M.; Use of computer in drug design and drug discovery: a review; International Journal of Pharmaceutical and Life sciences; 2012; 1(2); 1-21. [33] Sharma S.K., Sharma E. and Sharma Y.; A review: Recent computational approaches in Medicinal chemistry: Computer aided drug designing and delivery. The Pharma. Innovation Journal 2017; 6(5); 5-10. [34] Liao C., Sitzmann M., Pugliese A. and Nicklaus M.C.; Software and resources for computational medicinal chemistry; Future Med. Chem.; 2012; 3(8); 1057-1085. [35] An J., Lee D.C. and Law A.H.; A novel small-molecule inhibitor of the avian influenza H5N1 virus determined through computational screening against the neuraminidase; J. Med. Chem.; 2009; 52(9); 2667-2672. [36] Schames J.R., Henchman R.H., Siegel J.S., Sotriffer C.A., Ni H. and McCammon J.A.; Discovery of a novel binding trench in HIV integrase; J. Med. Chem.; 2004; 47(8); 1879-1881. [37] 120. Levin J.I., Chen J.M. and Laakso L.M.; Acetylenic TACE inhibitors. Part 3: Thiomorpholine sulphonamide hydroxamates; Bioorg. Med. Chem. Lett.; 2006; 16(6); 1605-1609. [38] Cohen N.C.; Structure-based drug design and the discovery of aliskiren (Tekturna): perseverance and creativity to overcome a R&D pipeline challenge; Chem. Biol. Drug Des.; 2007; 70(6); 557-565. [39] Morris G.M., Goodsell D.S. and Halliday R.S.; Automated docking using a lamarckian genetic algorithm and an empirical binding free energy function; J. Comput. Chem.; 2012; 19(14); 1639-1662. [40] Eccles S.A., Massey A. and Raynaud F.I.; NVP-AUY922: A novel heat shock protein 90 inhibitor active against xenograft tumor growth, angiogenesis, and metastasis; Cancer Res.; 2008; 68(8); 2850-2860. [41] Kim C.U., Lew W. and Williams M.A.; Influenza neuraminidase inhibitors possessing a novel hydrophobic interaction in the enzyme active site: Design, synthesis and structural analysis of carbocyclicsialic acid analogues with potent anti-influenza activity; J. Am. Chem. Soc.; 1997; 119(4); 681-690. [42] He L.W., Dai W.C. and Li N.G.; Development of orally active thrombin inhibitors for the treatment of thrombotic disorder diseases; Molecules; 2015; 20(6); 11046-11062. [43] Du H., Li J., Cai Y., Zhang H., Liu G., Tang Y. and Li W.; Computational investigation of ligand binding to the peripheral site in CYP3A4: Conformational dynamics and inhibitor discovery; Journal of Chemical Information and Modelling; 2017; 57(3); 616-626. [44] Maia E.H., Campos V.A., Santos B.D.R., Costa M.S., Lima I.G., Greco S.J., Ribeiro R.I., Munayer F.M., Silva A.M.D. and Taranto A.G.; Octopus: a platform for the virtual high-throughput screening of a pool of compounds against a set of molecular targets; Journal of Molecular Modelling; 2017; 23(1); 26. [45] Du X., Li Y., Xia Y.L., Ai S.M., Liang J., Sang P., Ji X.L. and Liu S.Q.; Insights into protein-ligand interactions: Mechanisms, models, and methods; International Journal of Molecular Sciences; 2016; 17(2); 14. [46] Ericksen S.S., Wu H., Zhang H., Michael L.A., Newton M.A., Hoffmann F.M. and Wildman S.A.; Machine learning consensus scoring improves performance across targets in structure-based virtual screening; Journal of Chemical Information and Modelling; 2017; 57(7); 1579-1590. [47] Kamarulzaman E.E., Vanderesse R., Gazzali A.M., Barberi-Heyob M., Boura C., Frochot C., Shawkataly O., Aubry A. and Wahab H.A.; Molecular modelling, synthesis and biological evaluation of peptide inhibitors as anti-angiogenic agent targeting neuropilin-1 for anticancer application; Journal of Biomolecular Structure and Dynamics; 2017; 35(1); 26-45. [48] Borhani D.W. and Shaw D.E.; The future of molecular dynamics simulations in drug discovery; Journal of Computer-Aided Molecular Design; 2012; 26(1); 15-26. [49] Macalino S.J.Y., Gosu Y., Hong S. and Choi S.; Role of computer-aided drug design in modern drug discovery; Archives of Pharm. Res.; 2015; 38(9); 1686-1701. [50] Laeeq S., Sirbaiya A.K., Siddiqui H.H. and Zaidi S.M.H.; An overview of the computer aided drug designing; World Journal of Pharmacy and Pharmaceutical Sciences; 2014; 3(5); 963-994. [51] Moffat J.G., Vincent F., Lee J.A., Eder J. and Prunotto M.; Opportunities and challenges in phenotypic drug discovery: an industry perspective; Nature Reviews Drug Discovery; 2017; 16(8); 202. [52] Sali A., Matsumoto R., McNeil H.P., Karplus M. and Stevens R.L.; Three-dimensional models of four mouse mast cell chymases. Identification of proteoglycan binding regions and protease-specific antigenic epitopes; Journal of Biological Chemistry; 2013; 268(12); 9023-9034. [53] Xu L.Z, Sánchez R., Sali A. and Heintz N.; Ligand specificity of brain lipid-binding protein; Journal of Biological Chemistry; 1996; 271(40); 24711-24719. [54] Pelkmans L. and Laat B.D.; Antibodies against domain I of ?2-glycoprotein I: the one and only?; Lupus; 2012; 21(7); 769-772. [55] Ring C.S., Sun E., McKerrow J.H., Lee G.K., Rosenthal P.J., Kuntz I.D. and Cohen F.E.; Structure-based inhibitor design by using protein models for the development of antiparasitic agents; Proceedings of the National Academy of Sciences; 1993; 90(8); 3583-3587. [56] Ma X.H., Shi Z., Tan C., Jiang Y., Go M.L., Low B.C. and Chen Y.Z.; In silico Approaches to multi-target drug discovery; Pharmaceutical Research; 2010; 27(5); 739-749. [57] Ambure P. and Roy K.; CADD modelling of multi-target drugs against alzheimer\'s disease; Current Drug Targets; 2017; 18(5); 522-533. [58] Abdolmaleki A., Ghasemi J.B. and Ghasemi F.; Computer aided drug design for multi-target drug design: SAR/QSAR, molecular docking and pharmacophore methods; Current Drug Targets; 2017; 18(5); 556-575. [59] Boissel J.P., Lee W.R., Presnell S.R., Cohen F.E. and Bunn H.F.; Erythropoietin structure-function relationships. Mutant proteins that test a model of tertiary structure; Journal of Biological Chemistry; 2013; 268(21); 15983-15993. [60] Hoque I., Bhattacharya S. and Chatterjee A.; Quantitative structure-activity relationships (QSARs) modeling of anti-hypertensive activity of dichloroanilino imidazoline derivatives; American Journal of PharmTech Research; 2018; 8(3); 161-178. [61] Manju K., Gunjan P. and Anju G.; Review on introduction to molecular docking software technique in medicinal chemistry; International Journal of Drug Research and Technology; 2017; 2(2); 8. [62] Eweas A.F., Maghrabi I.A. and Namarneh A.I.; Advances in molecular modelling and docking as a tool for modern drug discovery; Der Pharma Chemica; 2014; 6(6); 211-228. [63] Dhamal K.S., Waghmare S.A., Kamble H., Shelar V.S. and Bhalekar O.S.; Role of computer aided drug design in drug discovery & development; International Journal of Research in Engineering and Science; 2022; 10(3); 1-5. [64] Kawade D., Kinkar M.G., Gore N., Gadodiya M., Sahastrabuddhe M. and Dubey M.; Computer-aided drug design (CADD) & molecular modelling importance in pharmaceutical sciences; Journal of Emerging Technologies and Innovative Research; 2021; 8(6); a575-a591. [65] Lipinski C.A.; Lead and drug-like compounds: the Rule-of-five revolution; Drug Discovery Today Technologies; 2004; 1(4); 337–341.
Copyright
Copyright © 2022 Rainul Islam, Sumit Maji, Souparna Kabiraj, Umme Habiba, Rohan Pal, Somenath Bhattacharya, Soumallya Chakraborty, Dr. Arin Bhattacharjee. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.

Download Paper
Paper Id : IJRASET42836
Publish Date : 2022-05-17
ISSN : 2321-9653
Publisher Name : IJRASET
DOI Link : Click Here
 Submit Paper Online
Submit Paper Online